MALEIC ANHYDRIDE
| Method no.: | 86 |
| Matrix: | Air |
| Target concentration: | 0.25 ppm (1 mg/m3) |
| Procedure: | Samples are collected by drawing air through glass fiber filters
coated with 2 mg of |
| Recommended air volume and sampling rate: |
60 L at 0.5 L/min |
| Reliable quantitation limit: | 8.3 ppb (33 µg/m3) |
| Standard error of estimate at the target concentration: (Section 4.7.) |
8.86% |
| Special requirement: | Submit the samples for analysis as soon as possible after sampling. If delay is unavoidable, store the samples in a refrigerator. Store samples in a refrigerator upon receipt at the laboratory. |
| Status of method: | Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. |
| Date: December 1990 | Chemist: Yihlin Chan |
Organic Methods Evaluation Branch
OSHA
Analytical Laboratory
Salt Lake City, Utah
1. General Discussion
1.1. Background
1.1.1. History
In OSHA Method 25 (Ref.
5.1.), maleic anhydride is collected and derivatized on
1.1.2. Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy.)
Inhalation of subacute levels of maleic anhydride can cause severe headaches, nosebleeds, nausea, and temporary impairment of vision. Exposure to maleic anhydride can also lead to conjunctivitis and corneal erosion. Repeated exposure to concentrations above 1.25 ppm has caused asthmatic responses in workers. Allergies have developed so that lower concentrations of maleic anhydride can no longer be tolerated. An increased incidence of bronchitis and dermatitis has also been noted among workers with long-term exposure to maleic anhydride. One case of pulmonary edema in a worker was reported. (Ref. 5.6.)
1.1.3. Workplace exposure
Through Diels-Alder syntheses and co-polymerization reactions,
maleic anhydride is used in the manufacture of
According to the 1972 NIOSH National Occupational Exposure Surveys, exposure tomaleic anhydride was noted in the following industries: nonmetallic mineral products (SIC code 3299), percent of employee exposed, 30.2%; terrazzo, tile, marble, mosaic (1743), 19.4%; printing ink (2893), 19.1%; paints and allied products (2851), 16.0%; transportation equipment and supplies (5088), 14.3%; bags other than textile bags (2643), 8.6%; and metal cans (3411), 6.9%. The 1982 NIOSH surveys found 28.1% of the employee in the adhesives and sealants (SIC code 2891) exposed to maleic anhydride. (Ref. 5.8.)
1.1.4. Physical properties and other descriptive information (Ref. 5.9. unless noted otherwise)
| chemical name: | maleic anhydride |
| CAS no.: | 108-31-6 |
| synonyms: | 2,5-furandione; cis-butenedioic anhydride;
|
| structure: | 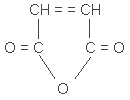 |
| mol wt: | 98.06 |
| boiling point: | 202°C |
| melting point: | 52.8°C |
| vapor pressure: | <21.3 Pa (0.16 mmHg) at 25°C |
| flash point: | 103°C |
| color: | colorless |
| solubility: | soluble in water, acetone, alcohol, and dioxane; partially soluble in chloroform and benzene |
| Derivative (Ref. 5.10.) | |
| structure: | Figure 1.1.4. |
| mol wt: | 265.27 |
| appearance: | white crystalline solid |
| melting point: | 146.0-146.5°C |
| solubility: | soluble in chloroform, methanol, acetonitrile, dimethylsulfoxide (DMSO). |
| UV spectrum: | Figure 1.1.4. |
The analyte air concentrations throughout this method are based on the recommended sampling and analytical parameters. Air concentrations listed in ppm are referenced to 25°C and 101.3 kPa (760 mmHg). The analyte concentrations are listed as those of maleic anhydride even though the derivative is the actual species analyzed.
1.2. Limit defining parameters
1.2.1. Detection limit of the analytical procedure
The detection limit of the analytical procedure is 7.1 ng per injection (15 µL injection of 0.471 µg/mL solution). This is the amount of analyte which gave a peak whose height is approximately 5 times the baseline noise. (Section 4.1.)
1.2.2. Detection limit of the overall procedure
The detection limit of the overall procedure is 1.99 µg per sample (8.3 ppb, 33 µg/m3). This is the amount of analyte spiked on the sampling device which allows recovery of an amount equivalent to the detection limit of the analytical procedure. (Section 4.2.)
1.2.3. Reliable quantitation limit
The reliable quantitation limit is 1.99 µg per sample (8.3 ppb, 33 µg/m3). This is the smallest amount of analyte spiked on the sampling device which can be quantitated within the requirements of a recovery of at least 75% and a precision (±1.96 SD) of ±25% or better. (Section 4.3.)
The reliable quantitation limit and detection limits reported in the method are based upon optimization of the instrument for the smallest possible amount of the analyte. When the target concentration of the analyte is exceptionally higher than these limits, they may not be attainable at the routine operating parameters.
1.2.4. Instrument response to the analyte
The instrument response over the concentration range of 0.5 to 2 times the target concentration is linear. (Section 4.4.)
1.2.5. Recovery
The recovery of the maleic anhydride-veratrylamine derivative
(MAVA) from the samples used in a
1.2.6. Precision (analytical procedure only)
The pooled coefficient of variation obtained from replicate injections of analytical standards at 0.5, 1, and 2 times the target concentration is 0.0074. (Section 4.6.)
1.2.7. Precision (overall procedure)
The precision at the 95% confidence level for the refrigerated 15-day storage test is ±17.4%. (Section 4.7.) This includes an additional ±5% for pump error. The overall procedure must provide results at the target concentration that are ±25% or better at the 95% confidence level.
1.2.8. Reproducibility
A draft copy of this procedure and six samples collected from a controlled test atmosphere (80% RH) were given to a chemist unassociated with this evaluation. The samples were analyzed after having been stored in a refrigerator at 0°C for 4 days. None of the sample results differed from its theoretical value by more than the precision reported in Section 1.2.7. (Section 4.8.)
1.3. Advantages
1.3.1. The maleic anhydride is derivatized in situ eliminating the possibility of its being hydrolyzed during storage. Interference from maleic acid is also eliminated.
1.3.2. Because the derivatizing agent is not leached from the filter during sampling, there is no need for an additional sorbent tube downstream of the sampler.
1.4. Disadvantages
The sampling medium is not available commercially.
2. Sampling Procedure
2.1. Apparatus
2.1.1. A personal sampling pump that can be calibrated to within ±5% of the recommended flow rate with the sampling device in line.
2.1.2. A three-piece polystyrene cassette containing two glass fiber filters, each coated with 2 mg of veratrylamine. (Figure 2.1.2.) Coated filters are prepared by applying 0.5 mL of a solution of 4 mg/mL veratrylamine in methylene chloride to each glass fiber filter and allowing them to dry in a hood or under vacuum. Store the coated filters in a closed jar in a refrigerator and use them within a month after preparation.
2.2. Reagents
No reagent is required for sampling.
2.3. Sampling technique
2.3.1. Remove the end plugs from the inlet and the outlet of the sampler. Attach the sampler to the sampling pump with a piece of flexible tubing and place it in the worker's breathing zone.
2.3.2. Replace the end plugs after sampling. Seal the sample
2.3.3. Submit at least one blank with each set of samples. Handle the blank the same as the other samples except draw no air through it.
2.3.4. List any potential interferences on the sample data sheet.
2.3.5. Submit the samples to the laboratory for analysis as soon as possible after sampling. If delay is unavoidable, store the samples at reduced temperature.
2.4. Sampler capacity
The sampler capacity was evaluated with a test atmosphere (80% RH) at 1.9 times the target concentration. The sampler capacity exceeds 130 L. (Section 4.9.)
2.5. Extraction efficiency and stability of extracted samples (Section 4.10.)
2.5.1. The average extraction efficiency at the target concentration was essentially quantitative (97.8%).
2.5.2. Extracted samples remain stable for at least 3 days when stored at room temperature.
2.6. Recommended air volume and sampling rate
2.6.1. The recommended air volume is 60 L.
2.6.2. The recommended air sampling rate is 0.5 L/min.
2.7. Interferences (sampling)
Excessive amounts of compounds that can react with veratrylamine, such as isocyanates, acid chlorides, and anhydrides other than maleic, may reduce the sampler capacity by consuming part of the derivatizing agent.
2.8. Safety precautions (sampling)
Attach the sampling equipment to the worker in such a manner that it will not interfere with work performance or safety. Follow all safety practices applicable to the work area.
3. Analytical Procedure
3.1. Apparatus
3.1.1. An HPLC equipped with a UV detector. A Waters 600E pump, a 900 photodiode array detector, and a WISP autosampler were used in this evaluation.
3.1.2. An HPLC column capable of separating veratrylamine, MAVA,
and any interferences. An Alltech C8 column (4.6 mm × 25 cm,
3.1.3. An electronic integrator or other suitable means of
measuring detector response. A
3.1.4. Sample vials, 4-mL glass, with Teflon-lined septum caps. WISP vials were used in this evaluation.
3.1.5. Volumetric flasks and pipets.
3.1.6. A mechanical shaker. A Fisher Roto-Rack® was used in this evaluation.
3.2. Reagents
3.2.1. Maleic anhydride-veratrylamine derivative (MAVA). Synthesized as in Section 4.12.
3.2.2. Dimethyl sulfoxide (DMSO). Dimethyl sulfoxide was obtained from Baxter Burdick and Jackson.
3.2.3. Acetonitrile. Acetonitrile was obtained from Baxter Burdick and Jackson.
3.2.4. Extraction solvent, acetonitrile/DMSO 90:10 (v/v).
3.2.5. Phosphoric acid. Phosphoric acid was obtained from J T Baker.
3.2.6. Water, HPLC grade. The water was from an in-house
Millipore
3.3. Standard preparation
3.3.1. Prepare stock standards by weighing 10-20 mg of MAVA in
(MW maleic anhydride) / (MW MAVA) = 98.06 / 265.27 = 0.3697
3.3.2. Prepare analytical standards by further diluting the stock standards with the extraction solvent. An analytical standard of 15 µg/mL represents 1 times the target concentration.
3.3.3. Prepare a sufficient number of standards to generate calibration curves. Analytical standard concentrations must bracket sample concentrations.
3.4. Sample preparation
3.4.1. Transfer the front and the back filters to separate WISP
vials. This is best accomplished by
3.4.2. Add 4.0 mL of the extraction solvent to each vial.
3.4.3. Cap the vials and shake them on a mechanical shaker for 1 h.
3.5. Analysis
3.5.1. HPLC conditions
| column: | Alltech C8 (4.6 mm × 25 cm,
|
| eluent: | acetonitrile/water/phosphoric acid 30:70:0.1 (v/v/v) |
| flow rate: | 1.2 mL/min |
| injection volume: | 15 µL |
| retention time: | 6 min |
| chromatogram: | Figure 3.5.1. |
| UV detector: | 254 nm (This wavelength was used because it gave good resolution of analyte from other components and good sensitivity.) |
3.5.2. Measure detector response using a suitable method such as electronic integration.
3.5.3. Construct a calibration curve using an external standard method by plotting µg per sample versus detector response of standard injections.
3.6. Interferences (analytical)
3.6.1. Any compound that absorbs at 254 nm and has a similar retention time as MAVA is a potential interference. Generally, chromatographic conditions can be altered to separate an interference.
3.6.2. Retention time on a single column is not considered proof of chemical identity. Additional means of identification include: analysis using an alternate HPLC column, detection at another wavelength, and comparison of absorbance response ratios.
3.7. Calculations
The analyte amount for samples is obtained from the calibration curve in terms of micrograms per sample uncorrected for extraction efficiency. The analyte amount is corrected by subtracting the amount found in the blank. The air concentration is obtained by using the following equations.
| mg/m3 = | (µg/sample)
(liters of air sampled)(extraction efficiency) |
| ppm = | (mg/m3)(24.46)
(98.06) |
| where | 24.46 = | molar volume (liters) at 101.3 Pa (760 mmHg) and 25°C |
| 98.06 = | molecular weight of maleic anhydride |
3.8. Safety precautions (analytical)
Avoid skin contact and inhalation of all chemicals. Restrict the use of all chemicals to a fume hood when possible. Wear safety glasses and a lab coat at all times while in the lab area.
4. Backup Data
4.1. Detection limit of the analytical procedure
The detection limit of the analytical procedure is 7.1 ng
4.2. Detection limit of the overall procedure
The detection limit of the overall procedure is 1.99 µg per sample
(8.3 ppb, 33 µg/m3). This is the amount of
analyte spiked on the sampling device which allows recovery of an
amount equivalent to the detection limit of the analytical procedure.
Six
|
| ||
| sample number |
theoretical amount (µg) |
amount recovered (µg) |
|
| ||
| 1 | 1.99 | 1.86 |
| 2 | 1.99 | 2.11 |
| 3 | 1.99 | 1.72 |
| 4 | 1.99 | 1.68 |
| 5 | 1.99 | 2.26 |
| 6 | 1.99 | 1.86 |
|
| ||
4.3. Reliable quantitation limit
The reliable quantitation limit is also 1.99 g per sample (8.3 ppb, 33 µg/m3). This was derived from the samples and data of Table 4.2. Because the recovery was greater than 75% and the precision (±1.96 SD) was ±25% or better, the detection limit of the overall procedure and reliable quantitation limit are the same.
|
| ||
| % recovery | statistics | |
|
| ||
| 93.5 | ||
| 106.0 | 96.2% | |
| 86.4 | SD = | 11.4% |
| 84.4 | Precision = | ± (1.96)(11.4%) |
| 113.6 | = | ± 22.3% |
| 93.5 | ||
|
| ||
The instrument response to MAVA over the range of 0.5 to 2 times the target concentration is linear with a slope of 58352 area counts per microgram per sample. The responses to MAVA at 254 nm were determined by multiple injections of analytical standards. The data which are summarized in Table 4.4. are presented graphically in Figure 4.4.
|
| |||
| × target concn µg/sample |
0.5× 31.42 |
1× 62.48 |
2× 125.68 |
|
| |||
| area counts | 1876030 | 3686990 | 7353160 |
| 1879430 | 3715800 | 7349260 | |
| 1889640 | 3738940 | 7391220 | |
| 1845030 | 3678670 | 7361270 | |
| 1878340 | 3650810 | 7380100 | |
| 1856180 | 3724480 | 7385300 | |
| 1870775 | 3699282 | 7370052 | |
|
| |||
A test atmosphere (80% RH) containing 1.86
mg/m3 of maleic anhydride was prepared in a
vapor generator.
|
| ||||||||
| storage time (days) |
% recovery (ambient) |
% recovery (refrigerated) | ||||||
|
| ||||||||
| 0 | 93.9 | 95.3 | 96.6 | 93.9 | 95.3 | 96.6 | ||
| 0 | 103.4 | 95.9 | 114.9 | 103.4 | 95.9 | 114.9 | ||
| 4 | 91.3 | 93.1 | 94.5 | 105.3 | 108.3 | 111.1 | ||
| 7 | 77.8 | 95.9 | 84.5 | 94.9 | 98.1 | 99.0 | ||
| 11 | 74.5 | 83.7 | 82.9 | 106.6 | 113.0 | 93.5 | ||
| 13 | 73.3 | 84.6 | 66.8 | 95.2 | 98.5 | 92.6 | ||
| 15 | 80.9 | 64.4 | 72.4 | 101.3 | 107.3 | 90.9 | ||
|
| ||||||||
4.6. Precision (analytical procedure)
The precision of the analytical procedure is 0.0074. The precision of the analytical procedure is defined as the pooled coefficient of variation determined from multiple injections of analytical standards representing 0.5, 1, and 2 times the target concentration (Section 4.4.).
|
| |||
| × target concn | 0.5× | 1× | 2× |
|
| |||
| SD | 16682 | 32882 | 17756 |
| CV | 0.0089 | 0.0089 | 0.0024 |
|
| |||
4.7. Precision (overall procedure)
The precision of the overall procedure is determined from the storage data. The determination of the standard error of estimate (SEE) for a regression line plotted through the graphed storage data allows the inclusion of storage time as one of the factors affecting overall precision. The SEE is similar to the standard deviation except it is a measure of dispersion of data about a regression line instead of about a mean. It is determined with the following equation:
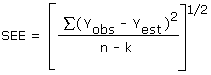
| where | n = k = k = |
total no. of data points 2 for linear regression 3 for quadratic regression |
| Yobs = | observed % recovery at a given time | |
| Yest = | estimated % recovery from the regression line at the same given time |
An additional ±5% for pump error is added to the SEE by the
addition of variances. The precision at the 95% confidence level is
obtained by multiplying the SEE (with pump error included) by 1.96
(the
Six samples, collected at 0.5 L/min for 60 minutes from a
controlled test atmosphere [80% RH,
|
| ||||
| sample no. | µg found | µg expected | % found | % deviation |
|
| ||||
| 1 | 58.584 | 63.24 | 92.6 | -7.4 |
| 2 | 58.748 | 62.97 | 93.3 | -6.7 |
| 3 | 66.636 | 62.90 | 105.9 | +5.9 |
| 4 | 60.100 | 62.17 | 96.7 | -3.3 |
| 5 | 61.832 | 64.90 | 95.3 | -4.7 |
| 6 | 63.840 | 62.99 | 101.3 | +1.3 |
|
| ||||
Sampler capacity was tested by sampling a test atmosphere of 1.9 mg/m3 maleic anhydride at ambient temperature and 80% relative humidity. Two samplers were placed in series. The upstream sampler contained only one filter. The back sampler was replaced with a new one every 40 minutes to monitor the downstream air concentration. The sampling rate was 0.501 L/min. Breakthrough was not seen in 280 minutes of testing.
|
| ||
| time (min) |
air volume (L) |
breakthrough (%) |
|
| ||
| 0- 40 | 20 | 0 |
| 40- 80 | 40 | 0 |
| 80-120 | 60 | 0 |
| 120-160 | 80 | 0 |
| 160-200 | 100 | 0 |
| 200-240 | 120 | 0 |
| 240-280 | 140 | 0 |
|
| ||
4.10. Extraction efficiency and stability of extracted samples
4.10.1. Extraction efficiency
The extraction efficiency for MAVA was determined by analyzing
the
|
| |||
| sample no. | µg spiked | µg recovered | % recovery |
|
| |||
| 1 | 59.0 | 56.36 | 95.5 |
| 2 | 59.0 | 57.09 | 96.8 |
| 3 | 59.0 | 57.74 | 97.9 |
| 4 | 59.0 | 57.32 | 97.2 |
| 5 | 59.0 | 59.05 | 100.1 |
| 6 | 59.0 | 58.72 | 99.5 |
| 97.8 | |||
|
| |||
4.10.2. Stability of the extracted samples
The stability of the extracted samples was investigated by reanalyzing the extracted samples with fresh standards 3 days after the original analysis. The samples had been recapped and stored at room temperature. The change in the recovery averaged -0.9%.
|
| ||
| initial recovery (%) |
recovery after 3 days (%) |
change (%) |
|
| ||
| 95.5 | 94.4 | -1.1 |
| 96.8 | 97.5 | +0.7 |
| 97.9 | 96.0 | -1.9 |
| 97.2 | 98.1 | +0.9 |
| 100.1 | 98.2 | -1.9 |
| 99.5 | 97.2 | -2.3 |
|
| ||
A chromatogram at the detection limit of the analytical procedure
is shown in Figure
4.1. and a chromatogram of one of the
4.12.1. Reagents
Veratrylamine, 97%, from Aldrich
Maleic
anhydride, from Aldrich
Toluene, from Baxter Burdick and
Jackson
Isooctane, from Fisher Scientific
Chloroform, from Fisher Scientific
4.12.2. Apparatus
Erlenmeyer flasks
Filtering flask
Fritted-glass filtering funnel
Explosion-proof hot
plate
4.12.3. Procedure
Recrystallize the maleic anhydride from toluene. Add 0.98 g (0.01
mole) of maleic anhydride to a solution of 1.67 g (0.01 mole) of
veratrylamine in 10 mL of chloroform. Stir the mixture for 10 min.
Evaporate the chloroform in a hood. Dissolve the residue in a
minimal amount of chloroform. While heating on a hot plate, slowly
add isooctane until the solution just becomes cloudy. Clear the
solution with an addition of a drop of chloroform. Remove from the
hot plate. After the solution has cooled to room temperature, set in
a freezer overnight. Collect the crystals that formed. The melting
point should be
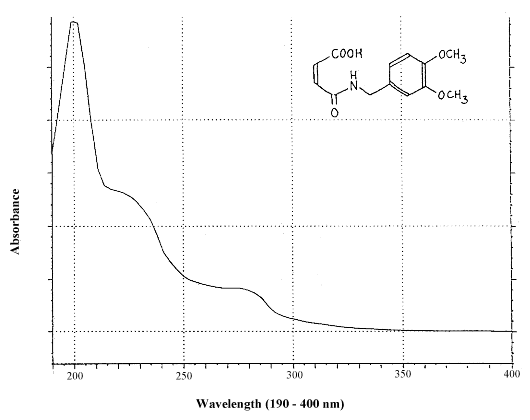
Figure 1.1.4. UV spectrum (in water/acetonitrile/phosphoric
acid = 70:30:0.1 (v/v/v)) and molecular structure of MAVA.
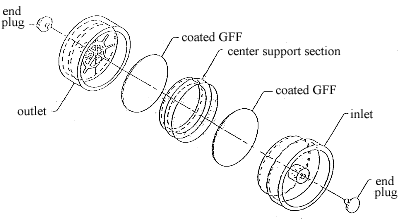
Figure 2.1.2. A drawing of a sampling device for maleic anhydride.
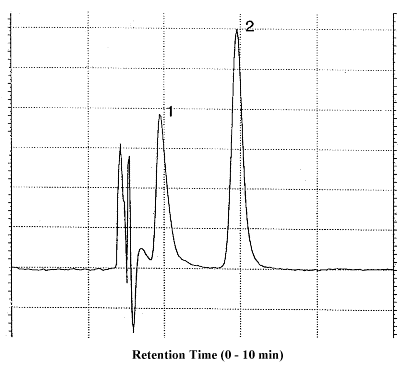
Figure 3.5.1. Chromatogram at target concentration.
1 = veratrylamine, 2 = MAVA.
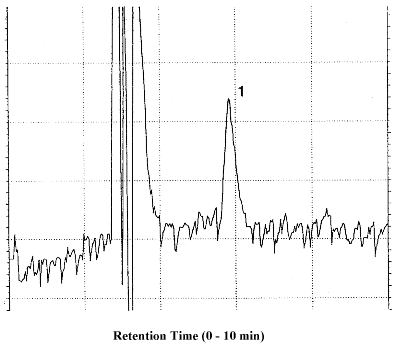
Figure 4.1. Detection
limit of the analytical procedure. 1 = MAVA. 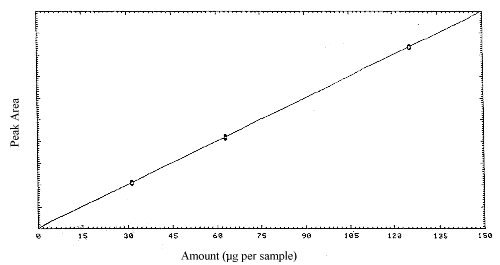
Figure 4.4. Calibration curve
for MAVA. 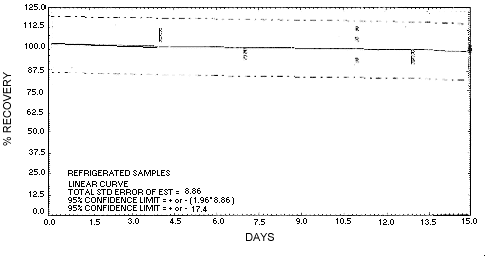
Figure 4.5.1. Storage test at reduced temperature.
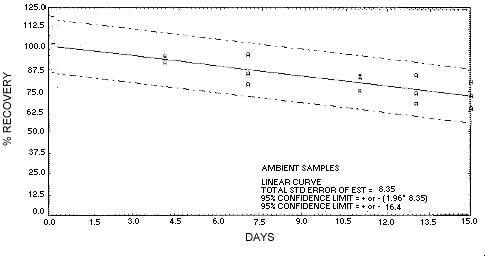
Figure 4.5.2. Storage test at ambient temperature.
5. References
5.1.
"OSHA Analytical Methods Manual", Second Edition, U.S. Department of Labor, Occupational Safety and Health Administration; OSHA Analytical Laboratory: Salt Lake City, UT, 1990; Method 25; American Conference of Governmental Industrial Hygienists (ACGIH): Cincinnati, OH, Publication No. 4542.5.2. ibid. Method 42.
5.3. ibid. Method 47.
5.4. ibid. Method 54.
5.5. Chan, Y., "OSHA Method 82: Acetic Anhydride", OSHA Analytical Laboratory, unpublished, Salt Lake City, UT 84165, April 1990.
5.6. Sittig, M., "Handbook of Toxic and Hazardous Chemicals", Second Edition, Noyes Publications, Park Ridge, NJ, 1985.
5.7. Budavari, S., Ed., "Merck Index", Eleventh Edition, Merck & Co., Rahway, NJ, 1989.
5.8. "OSHA Regulated Hazardous Substances,
Industrial Exposure and Control Technologies", U.S. Department of
Labor, Occupational Safety and Health Administration, Washington,
D.C., 1990, ISBN:
5.9. Sweet, D.V., Ed., "Registry of Toxic Effects
of Chemical Substances",
5.10. Author's personal observation.