QUALITATIVE X-RAY FLUORESCENCE ANALYSIS OF WORKPLACE SUBSTANCES
| Method Number | ID-204 |
| OSHA Permissible Exposure Limit (PEL) |
Provides qualitative element identification for the regulated substances listed in Section 4.1., Table 1. |
| Sampling Matrix | Air filter, wipe filter, and bulk material |
| Sampling Procedure | Samples are collected either as air samples on
|
| Air Volume | Obtain full work-shift air samples when possible. |
| Sampling Rate | 2 L/min for personal samples. If possible, take area samples at 9 L/min. |
| Analytical Procedure | All samples are analyzed with minimal sample preparation using
an Energy Dispersive |
| Qualitative Detection Limit | |
| Air Samples | Typically 0.1 to 30 µ. See Section 4.2., Table 2 for specific air sample detection limits. |
| Bulk Samples | Typically 0.01 to 8%. Potential |
| Status of Method | Evaluated qualitative method |
| Date | September, 1990 |
| Chemist | Mike C. Rose |
Commercial manufacturers and products mentioned in this method are for descriptive use only and do not constitute endorsements byUSDOL-OSHA. Similar products from other sources can be substituted.
OSHA Technical Center
Salt Lake City, Utah
1. Introduction
This method describes the sampling and semiquantitative
- 1.1. History
- 1.1.1. Previously, samples submitted to the OSHA Laboratory for
qualitation were analyzed manually using a Finnigan Model 8000
1.1.2. Neutron Activation Analysis was also used for element
identification. This analysis was
1.1.3. Inductively Coupled Plasma-Atomic Emission Spectroscopy
1.1.4. This method was evaluated using the OSHA Laboratory's XRF
system. It consisted of a Kevex 770
1.2. Principles
- 1.2.1.
In an EDXRF spectrometer,
The approximate relationship between an element's atomic number
and the energy of individual emission lines for each specific
Where:
E = energy of X
ray
a = proportionality
constant
Z = atomic
number
s =
constant for each line series
Moseley's law indicates that an element's spectral lines are a smooth function of the atomic number. The spectral lines for elements with low atomic number (light elements) occur at lower energies than the corresponding lines for elements with high atomic number (heavy elements). The peak energies and spectral group patterns provide for qualitative identification.
1.2.2. Data workup depends on the manner of sample preparation - thin films or thick dusts.
| a) | Thin films |
| For uniform thin deposits of material on a support
medium that is transparent to | |
| b) | For thick samples and powders consisting of a few grams of
material approximately a centimeter deep,
|
| c) | Non-linear calibration curves can also be used to correct
for other instrumental realities (e.g., fluorescing support
medium or |
1.2.3. The results from EDXRF analyses are used for analytical support and fit into the following scheme:
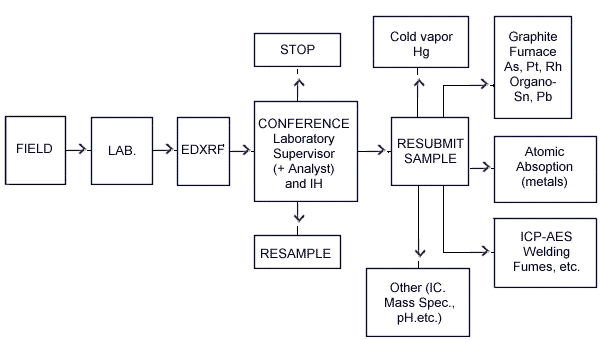
This approach screens air samples of unknown composition to identify elements in dusts listed in Section 4.1., Table 1. It is also used to make a semiquantitative determination of the composition of bulk samples. The information obtained during the screening is used to determine whether additional time and resources are necessary to quantitatively identify the constituents in bulk, wipe, or certain air samples. Samples analyzed by XRF take only minutes to prepare, are not destroyed in the process, and do not require analytical standards for each screening or semiquantitative determination.
1.3. Method Performance
The detection limits reported in this method are based upon the
optimization of the instrument for the maximum practical signal. The
microgram detection limits reported for air samples are for analyte
elements dispersed as aerosols concentrated near the center on the
surface of polyvinyl chloride (PVC) membranes. PVC membranes were
selected over
- 1.3.1. Analytical detection limit
Detection limits for filter samples are listed and discussed in Section 4.2., Table 2.
| a) | Aerosol samples | |
| The approach used to calculate detection limits is attributed to Birks (5.4.) and is given in Bertin (5.5.). The following equation (based on Poisson counting statistics) was used to estimate detection limits (DL): | ||
| DL = 3(A/C)(B)1/2 | ||
| Where: A = analyte mass, (µg) B = blank counts C = analyte counts | ||
| The blank counts were determined in the same energy region used for profile-fitting the analyte counts. The analyte counts were determined from a peak profile fit of either: | ||
| 1) The blank- and background-subtracted analyte
peak. 2) The background-subtracted analyte peak in cases where blank subtraction would yield negative counts. | ||
| For aerosol air samples collected on PVC
membranes, the detection limit ranged from about 30 µg for
elements with atomic numbers below 17 (chlorine) to less than
4 µg for elements with atomic numbers above 17. When
determining these detection limits (Section 4.2., Table 2),
| ||
| b) | For powdered bulk samples, matrix effects can
have a profound effect on the lower levels of
detection. A wide range of sample types was evaluated in the
bulk tests. Based on the data shown in Section 4.3., Tables
| |
1.3.2. Instrument response to the analyte
The instrument response is sample and matrix dependent. For air and bulk samples, the lower qualitative limit is the detection limit. For homogeneous powdered bulk samples, the semiquantitative working range extends from the detection limit to near 100% of an analyte.
1.3.3. Recovery
Recoveries are matrix dependent. Typical recoveries for elements
in powdered bulk samples are listed in Section 4.3., Tables
1.4. Advantages
Provides rapid, non-destructive analyses
Affords qualitative
information for a large number of elements
Can be
semiquantitative
Can identify unexpected elements
Requires no
sampling reagents
1.5. Disadvantages
Analysis requires expensive instrumentation and support
software
Requires experienced analyst(s)
Limited use in
quantitative analysis
Analysis is matrix dependent
Requires
information about the sample matrix, chemistry, and suspected elements
to achieve the most accurate analysis
2. Sampling
- 2.1. Safety Precautions
- 2.1.1. Attach the sampling equipment to the worker such that it
will not interfere with work performance or safety.
2.1.2. Follow all safety practices that apply to the work area being sampled.
2.2. Equipment
- 2.2.1. Air sampling
| a) | Mixed-cellulose ester (MCE) filters, 0.8-µm pore size,
cellulose backup pads, and cassettes, |
| b) | Low-ash PVC membrane filter (use for gravimetric
determinations or when quartz determinations are necessary),
|
| c) | Cellulose back-up pads (support pads) (MSA, Pittsburgh, PA). |
| d) | Clear polystyrene, 37-mm inside diameter, closed-face
cassette, (two-section, SKC part no. 225-2 or
|
| e) | Gel bands (Omega Specialty Instrument Co., Chelmsford, MA) for sealing cassettes. |
| f) | Sampling pump Personal samples: Use a personal sampling pump that can be calibrated to within ±5% of 2; L/min with the sampling device attached. Area samples: Use a higher volume sampling pump capable of 5 to 9 L/min. |
| g) | Cyclone (only if respirable dust sampling is necessary);
Nylon, |
| h) | Assorted flexible tubing |
| i) | Stopwatch and bubble tube or meter for pump calibration |
| j) | Analytical balance (0.01 mg). |
| k) | Desiccant (Drierite or similar material) and desiccating chamber. (Note: Use only if weights of air samples are desired). |
2.2.2. Bulk sampling
| a) | Scintillation vials, 20-mL, (part no. 74515 or 58515,
Kimble, Div. of |
2.2.3. Wipe sampling
| (Note: | Wipe samples are not an optimum medium for this method - See Section 2.2.3. for further details.) |
| a) | Smear tabs (part no. 225-24, SKC Inc., Eighty Four, PA, or Whatman no. 41 or no. 42 filters, Whatman LabSales Inc., Hillsboro, OR). Filters composed of PVC or MCE (Section 2.2.1.) can also be used to take wipe samples. |
| b) | Scintillation vials, 20-mL (as described above). |
2.3. Sampling Techniques
See Section 4.1., Table 1 for additional sampling information regarding substances having specific dust PELs.
- 2.3.1. Air sample collection
If sample weights are of interest, desiccate and then weigh any PVC filters before sampling.
Due to the nature of substances collected and analyzed using
this method, it is recommended that samples taken for compliance
purposes are pre- and
For XRF analyses, MCE filters are preferred over PVC because they are more transparent to X rays and blank intensities are less significant. However, sample weights are better determined using the PVC filter because moisture retention is minimal. Use PVC membrane filters for gravimetric analyses.
| 1) | Place a cellulose backup pad in a cassette. Place the
membrane filter (either MCE or PVC) on top of the backup pad.
If large loadings are expected and the membrane has a smooth
and a rough side, place the membrane in the cassette with the
smooth side against the backup pad and use a
|
| 2) | Attach a Tygon tube between the pump and a flow calibration cassette so that the air will be drawn through the filter membrane. Do not place any tubing in front of the cassette. |
| 3) | Calibrate each sampling pump to within ±5% of the
recommended sampling rate with the calibration cassette
attached |
| 4) | Attach a prepared cassette to the calibrated sampling pump and place in the employee's breathing zone. |
| 5) | If possible, take a full shift sample at the recommended sampling rate. |
| 6) | Place plastic end caps on each cassette after sampling. |
| 7) | If weights are of interest, remove any PVC filters from
the cassettes, dessicate, and then |
| 8) | Attach an OSHA-21 seal around each air and blank sample in such a way as to secure the end caps of the cassettes. |
| 9) | Submit at least one blank sample with each set of air samples. |
| 10) | Gravimetric analyses in the field should suffice when the mg/m3 respirable dust PEL for a substance is evaluated. Any respirable dust samples suspected of containing quartz should be submitted to the laboratory for quartz analysis. Also, situations may arise where the IH needs further information to characterize a respirable dust exposure. In these cases, respirable dust samples can be submitted for laboratory analysis. |
2.3.2. Bulk sample collection
In order of laboratory preference, bulk samples may be one of the following:
- a) a high-volume filter sample,
b) a representative settled dust (rafter) sample,
c) a sample of homogeneous dust (or powdered) bulk material in the workplace.
| 1) | Collect between 10 to 20 mL of dry bulk sample to provide
for optimum detection of minor components in bulk samples.
Samples of at least |
| 2) | Transfer the bulk material into a 20-mL scintillation
vial, seal with a cap having an inert plastic liner, and wrap
with vinyl or electrical tape. Securely wrap an
|
| 3) | The type of bulk sample should be stated on the OSHA 91
and |
2.3.3. Wipe sample collection
Wipe samples are not an optimum medium for this method; increased background signal noise results in high detection limits and irreproducible blank corrections. Substances collected on wipes are unevenly distributed. If necessary, qualitative scans of a portion of the wipe sample can be performed.
| 1) | Wear clean, impervious, disposable gloves when taking each wipe sample. |
| 2) | Moisten the wipe filters with deionized water prior to use. |
| 3) | If possible, wipe a surface area covering 100 cm2. |
| 4) | Fold the wipe sample with the exposed side in. |
| 5) | Transfer the wipe sample into a 20-mL scintillation vial,
seal with a cap having an inert plastic liner, and wrap with
vinyl or electrical tape. Securely wrap an
|
2.4. Sample Shipment
- 2.4.1. Document the operation and indicate any known or
suspected elements and compounds. If possible, indicate whether
components that volatilize may be present.
Any information regarding suspected sample composition, industrial operation, etc. will aid in obtaining the most accurate analysis. These details can assist the analyst when optimizing the instrument and call attention to potential interferences.
2.4.2. Request QUAL-XRF analysis and any appropriate follow-up quantitative analysis.
2.4.3. Ship air and blank samples to the laboratory with appropriate paperwork.
2.4.4. Bulk and wipe samples should be shipped separately from air samples. They should be accompanied by Material Safety Data Sheets (MSDS) if available. Check current shipping restrictions and ship to the laboratory by the appropriate method.
3. Analysis
The user must decide upon the applicability of available equipment
and software when using this method. This method is performed using an
EDXRF; however, the analyses can be conducted using wavelength dispersive
- 3.1. Safety Precautions
- 3.1.1. Chemical
Handle reagents and bulk samples carefully. Use protective
equipment such as: Gloves, laboratory coats, safety glasses, and an
exhaust hood. Use a
3.1.2. Radiation
| a) | When samples are suspected of containing
|
| b) | Follow established laboratory safety guidelines. Modern
WARNING: These devices should not be adjusted, removed, or overridden for any reason. |
| c) | Radiation monitors are worn by |
| d) | There should be a red or yellow warning light which, when
lit, indicates the |
| e) | Periodically have safety mechanisms checked to determine
satisfactory operation. A sensitive,
|
| f) | Avoid inserting fingers into the sample compartment. Use forceps to change samples. |
3.2. Equipment
- 3.2.1.
The spectrometer should be equipped with appropriate monitors, collimators, and secondary targets. The spectrometer at the OSHA Laboratory included the following:
- Lucite monitor
Tantalum collimator
Gadolinium secondary target with gadolinium filter
Silver secondary target with silver filter
Zirconium secondary target with zirconium filter
Germanium secondary target
Titanium secondary target
3.2.2. Sample holders for cups
3.2.3. Sample holders for air filters
3.2.4. Sample cups
3.2.5. Kapton window film, 0.33 mil thick (part no. 3511, SPEX Industries, Edison, NJ)
3.2.6. Mylar window film, 0.25 mil thick (part no. 3517, SPEX Industries)
3.2.7. Mylar window film, 0.14 mil thick Ultra-thin Mylar, (part no. D12-202, Kevex Corporation, San Carlos, CA)
3.2.8. Polypropylene window film, 0.20 mil thick (part no. 3520, SPEX Industries)
3.2.9. Microporous window film, polypropylene (part no. D12-203, Kevex Corporation)
3.2.10. Radiation safety monitor (model Monitor 4, S.E. International Instrumentation Division, Summertown, TN)
3.2.11. Platform balance capable of 0.01 g precision and at least 50 g range
3.2.12. Vacuum desiccator - use for sample preparation (model no.
F42020,
3.2.13. Vacuum pump - use for sample preparation (model no. DD 20, Precision Scientific, Chicago, IL)
3.3. Reagents (use reagent grade or better powders for calibrations).
- 3.3.1. Boric acid
3.3.2. Graphite
3.3.3. Sodium bicarbonate
3.3.4. Aluminum oxide
3.3.5. Ammonium sulfate
3.3.6. Titanium dioxide
3.3.7. Zinc oxide
3.3.8. Yttrium oxide
3.3.9. Aluminum sheet, 1 mm thick
3.3.10. Copper sheet, 1 mm thick
3.4. Instrument Calibration
This method is optimized for the analysis of powdered bulk samples. Use appropriate materials and manufacturer recommendations when calibrating specific instrumentation and software. For the purposes of this method, calibration Sections 3.4.2. to 3.4.5. should be performed only once for a properly maintained instrument. Examples of the calibrations performed on the equipment described above are given in the Standard Operating Procedure (SOP) (5.6.) and in Section 4.4., Table 5a.
- 3.4.1. Prepare appropriate standard(s) and perform an energy
calibration of the EDXRF spectrometer.
3.4.2. Determine the peak-width at half-maximum for calibrating the peak deconvolution (profile fitting) software. (This is typically performed when the instrument is installed and then checked periodically during preventive maintenance.)
3.4.3. If necessary, calibrate the instrument for fundamental
3.4.4. Calibrate the instrument for light element corrections. For example, the following powder samples might be selected and prepared as bulks in appropriate sample holders:
- Graphite
Boric acid
Sodium bicarbonate
Ammonium sulfate
Aluminum oxide
When obtaining scatter data, use an energy scale range
appropriate to include the
3.4.5. Run a variety of known powdered materials and perform adjustments as necessary to improve recoveries.
3.5. Sample Preparation
Check the sample documentation for information regarding composition. Knowledge of the composition provides a basis for handling potential interferences and assists in selecting the appropriate computer model to account for any matrix effects.
Perform assembly of sample holders on a clean dust-free surface. Use sample holders appropriate for the instrument. (Note: The instrument mentioned in the method and evaluation had the following sample/detector/target geometry: The analytical surface is horizontal to and above the detector and target. Samples placed "dust side down" are placed with the dust side oriented towards the target and detector.)
- 3.5.1. Air sample preparation - MCE and PVC filters
| 1) | Decide how to present the sample for analysis. | |
| a) | Filters with ADHERENT DUST are
| |
| b) | Loose dust on filters can be analyzed dust side up, but only if great care is taken. There is a potential for contaminating the sample chamber. | |
| 2) | Assemble the filter holders. The air sample holders used in the evaluation of this method are shown below. | |
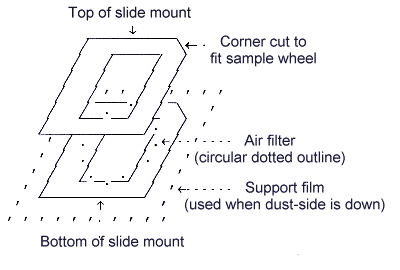
Air sample mount used on Kevex 8000/770
3.5.2. Bulk samples
Samples in the liquid state are generally not analyzed. The
liquid phase can be evaporated and the
| 1) | Film support selection For this method, bulk samples may be analyzed on 0.14-mil (3.6-µ) Mylar film. Other materials are available and can be used if samples are chemically incompatible with Mylar, but light element recoveries and Compton to Rayleigh scatter ratio data will be affected. These materials and compatibilities are more fully described in the SOP (5.6.). |
| 2) | The bulk sample holders used during the evaluation of this method are shown below. |
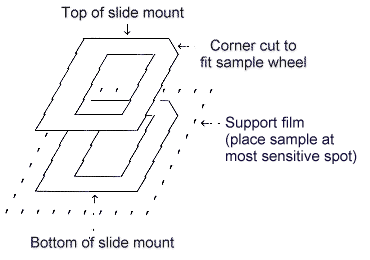
Bulk sample mount used on Kevex 8000/700
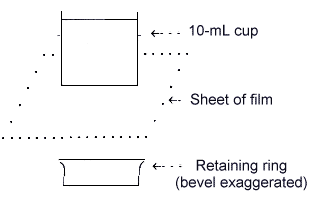
Sample cup used for powdered bulk samples
(Optional Microporous film may be used with an
additional retaining ring on top to cover the sample.)
| a) | Liquid bulk or small amounts of dry bulk
samples A qualitative analysis should only be performed if a sample consists of: evaporated deposits | |||
| An attempt should be made to prepare this type
of sample as a thin, even layer on the support film. This
reduces sample matrix effects; however, increased detection
limits due to decreased sensitivity are noted. When a sample
cannot be spread evenly, position the sample at the most
sensitive location on the sample holder. This location can be
determined by trial and error using copper peak intensities
from a small ring of fine copper wire and a sample holder
containing a support film. Mark the location of the ring
center on the support film with a | ||||
| Liquid bulk or small amounts of dry bulk samples are prepared by the following procedure: | ||||
| 1) | Select a film material chemically compatible with the sample. The films most often used are made of Mylar, Polypropylene, or Kapton. Further information regarding specific incompatibilities is listed in the SOP (5.6.) and manufacturer catalogs. | |||
| 2) | Assemble the sample holder. | |||
| 3) | Position a small volume of the powdered bulk specimen or several drops of liquid sample at the most analytically sensitive location on the film. For liquid samples, place the film holding the liquid sample in a vacuum desiccator with a liquid nitrogen trap to catch vapors. Evaporate the liquid to dryness and then slowly let air into the desiccator so as not to disturb the dried material. Some oxidizing agents or organic substances may attack all three films mentioned above. For this reason, it is important to reduce the time that solvents are in contact with the film; therefore, begin evaporation as soon as possible after spotting the film. Substances such as sulfuric acid and sodium hydroxide become more concentrated and reactive after evaporation. Ammonium carbonate or boric acid can be added to neutralize acids or bases respectively. If not neutralized, a rapid analysis and removal from the sample chamber is desirable. | |||
| b) | Large quantities of bulk dust (thick) samples If a sufficient amount (> 0.5 g) of finely powdered dust is available, a semiquantitative analysis can be performed. These bulks are best presented as a thick layer of dust in a sample cup. This greatly improves detection limits and minor component identifications; however, increased matrix effects are also noted. The sample should be homogeneous because the entire contents of the cup are not analyzed. | |||
| 1) | Assemble bulk sample cups and place in sample holders. Use
| |||
| 2) | If manufacturer software requires sample mass thickness
data(mg/cm²), perform the following: Tare the sample cup on a balance capable of 0.01 g precision. Pour some of the powdered bulk into the cup until the depth reaches 1 to 2 cm (approximately 5 mL). Record the weight of the powder. Calculate the sample mass thickness by dividing the sample mass (in mg) by the area (in cm²). To obtain the mass thickness for samples contained in
Record the mass thickness for each sample. | |||
| 3) | If it is necessary to perform light element analyses on dusty bulks, protect the instrument sample chamber and vacuum pump from dust cloud contamination by either sealing the top of the sample cups with Microporous polypropylene film (using a retaining ring) or by substituting He for the vacuum. Coal dust is a common example of a dust that tends to form a dust cloud when a vacuum is drawn. Check for potential dust cloud generation by first subjecting each sample to a vacuum in the vacuum desiccator. | |||
| 4) | For bulk blanks, use an air filter sample holder to analyze the support medium used in the assembly of the sample cup. This is performed in order to avoid detecting scattered and fluoresced radiation from an empty bulk sample cup. (Normally when analyzing bulk material, the sample cup walls are blocked by the sample.) | |||
3.6. Analysis
- 3.6.1. Analytical conditions
Use
3.6.2. Desirable analyte sensitivities
See Section 4.2., Table 2 and Section 4.4., Table 5a for examples of integrated peak areas obtained using the instrumentation specified in Section 1.1.4.
3.7. Interferences
- 3.7.1. Positive interferences (non-analyte signal-augmenting
phenomena) include background signals; instrument artifacts from
electronics, collimators, target, and filter fluorescence; target
and filter Compton and Rayleigh scatter peaks; escape peaks; sum
peaks; overlapping sets of M, L, and K spectral lines (MLK peaks)
from elements other than those of interest; matrix specific
enhancement; and closer sample placement. Many interferences can
be resolved through software, by blank subtraction, or by
identification of blank contaminants. Sum and escape peaks are
further discussed:
| a) | Sum peaks occur when more than one photon arrive
coincident at the detector. The problem of sum peaks can be
reduced by decreasing the |
| b) | An escape peak is generated by the low-probability quantum excitation of the K-shell electrons in the silicon atoms of the detector producing a small peak at 1.76 thousand electron volts (kV) below a fluorescence line. Fluorescence lines below 1.76 kV are unaffected, whereas those just above 1.76 kV are most strongly affected. This phenomenon is easily modeled, so software can readily correct for it. |
Alternative analytical lines are often available to resolve interferences.
3.7.2. Negative interferences (signal-decreasing phenomena) include matrix absorption effects and displacement of the sample away from the secondary target and detector. Matrix absorption effects can be addressed using sample information provided by the IH. Sample displacement errors can be reduced by using care to prepare flat membrane support surfaces.
3.7.3. Peak location in a spectrum is not proof of the identity of an element. Analysis of other peaks for that element and profile fitting (also called deconvolution), if necessary, provide further evidence of identity. Qualitative analysis requires experience and analyst interaction.
3.8. Calculations
The sequence of steps in evaluating the data depends on software
requirements. Alternate sequences may be necessary when using
different software. The steps below assume certain software features
are available to the user. Other software products may be used.
Qualitative analysis consists of Sections
- 3.8.1. Perform escape peak corrections.
3.8.2. Perform sum peak corrections, if available.
3.8.3. Perform blank corrections for membrane support (or air blank).
3.8.4. Perform automated identification of elements. Note: This is an optional step. Automated identification may suggest possible elements that the analyst may not have considered.
3.8.5. Perform background modeling and subtraction.
3.8.6. Identify the elements and interferences present using the systems graphic terminal and peak markers (which indicate MLK spectral locations). However, neither automated identification nor a trained analyst may be able to identify elements whose major peaks occur as shoulders on the peaks of other elements present in the matrix. When characterizing a sample, also consider the particular elements indicated on the sample documentation. Input the identified elements into the software.
3.8.7. Deconvolute (profile-fit) the identified elements to obtain integrated (area) counts for the analytical peaks.
3.8.8. Check for residual peaks. Uncorrected sum peaks and the peaks of unidentified elements may remain. This is an opportunity to identify elements that are subject to significant interferences, e.g., analyte peaks that occur only as shoulders on the peaks of other elements in a particular matrix. Repeat Sections 3.8.6. and 3.8.7. until all peaks are accounted for.
3.8.9. Determine the Compton to Rayleigh scatter ratio.
3.8.10. Perform the fundamental parameters estimation including the sample mass thickness and Compton to Rayleigh scatter data. [Note: This latter approach is especially useful when analyzing light matrices.]
3.8.11. Repeat 3.8.10. without the Compton to Rayleigh scatter data. [Note: This approach is useful when the sample matrix is unknown.]
3.8.12. Repeat 3.8.11. and force the results to total 100%. This approach is useful when all major elements in the sample have been accounted for.
3.8.13. Include any known (or suspected) chemistry (e.g., whether the sample consists of geological material, oxides, sulfides, alloys, organic, or other light element composition). Also include any known chemical stoichiometry of the analyzed elements to help account for unanalyzed elements such as the light elements Na, O, C, and H. Chemistry information places constraints on how the results are calculated and generally improves the reliability of the semiquantitative estimates. For example, for many mineral dusts, it may be appropriate to represent the analyzed elements as oxide compounds such as Fe2O3, TiO2, SiO2, CaO (or CaCO3), and BaO. More specific knowledge about the matrix may be used. For example, if a sample theoretically consists of primarily anhydrous sodium sulfate and sodium chloride, represent the analyzed elements S and Cl as Na2SO4 and NaCl to account for the unanalyzed Na and O contents. Repeat Sections 3.8.10. through 3.8.12. to include the chemistry constraints.
3.8.14. The semiquantitative results from the operations above may differ significantly. Analyst experience and matrix information provided about the sample must be used to select the results that represent the most realistic physical and chemical assessment.
3.8.15. Re-analyze at least 10% of the samples submitted for
semiquantitative XRF analysis by validated
3.9. Reporting Results
Results for the following samples are generally reported as qualitative:
- Air or wipe filter samples
Liquid bulk samples
Insufficient amount of bulk material (usually <0.5 g)
- 3.9.1. Qualitative results
Report the elements identified by XRF analysis using element symbols. Rank the element symbols based on atomic number without regard to amounts.
The element symbols may be further qualified as follows:
| 1) | "+" to indicate detected and confirmed present (e.g., "+ Fe") |
| 2) | "-" to indicate that a requested analyte was specifically looked for, but was not detected (e.g., "- Br") |
| 3) | "?" to indicate that a signal was present indicating that the element may be present, but it could not be confirmed on alternate peaks in this matrix (e.g., "? As" in a matrix containing Pb) |
All of the identified elements need not be reported. Unreported elements may include those near the detection limit or those having significant interferences on all major analytical lines.
3.9.2. Semiquantitative results
All semiquantitative results are approximate. It is important to consider the limited accuracy of this method. The method evaluation indicated that errors in quantitation by a factor of 2 are not uncommon. Additional work can be performed to improve analytical results and some suggestions are mentioned in the Appendix.
Semiquantitative results may be reported two different ways:
| a) | In cases where samples were analyzed as homogeneous powders of uniform thickness, rank the element symbols (and qualifiers) from highest to lowest estimated concentration. This is the most restrained (or conservative) representation of the semiquantitative information. |
| b) | Numerical semiquantitative results (with units of "%" or "µ/g") can be added to the list of identified substances in Section 3.9.2.a. Although reported to two significant figures, these results should be considered as "order of magnitude" estimates. |
3.9.3. When routing samples for re-analysis by another method,
include a copy of the semiquantitative numerical results. While
not as detailed as an MSDS, these results provide useful
information to those who must handle the bulk. Results also assist
in bulk sample preparation to select both appropriate digestion
techniques and aliquot sizes. Also request that the results
obtained by the
4. Backup Data
An evaluation of this method was conducted to address qualitative
support for aerosol (air) and bulk samples, and the potential for
analyzing bulk materials semi-quantitatively without the use of specific
calibration standards. Samples were prepared and analyzed during this
evaluation as described in Section 3. of the method. Fourteen air
samples on PVC and
- 4.1.
PELs Supported
Table 1 (Regulated Dusts)
4.2. Estimation of Aerosol Detection Limits
Experimental design
- Table
2a (Aerosol Source Materials)
Table 2b (Estimated Detection Limits)
Table 2c (Estimated Aerosol Detection Limits - Conservative)
Discussion of aerosol detection limits
4.3. Evaluation - Bulk Sample Determinations
Experimental design
Calculations used in software
- Table
3 [Pure Substances -
(NH4)2SO4
and
Al2O3]
Table 4a (Homogeneous Light Element Matrices - TEG50-B and TEG50-C)
Table 4b (Heterogeneous Intermediate Matrices - NIST SRMs 635, 636, 637, 1881, and 2704)
Table 4c (Heterogeneous Mixed Matrix Types - V1 through V12)
Table 4d (Potential Worst-Case Bulk Detection Limits)
4.4. Kevex Operating Conditions Used in Evaluations
Experimental design 4.5. Conclusions
4.6. Appendix
Additional recommendations to improve aerosol detection limits
Additional recommendations to improve semiquantitative estimates
4.1. PELs Supported (Back to Outline)
Listed below are those compounds that may be characterized using this method; however, when the analysis of a specific compound is requested, an elemental analysis is performed and reported as the compound.
| Table 1 Regulated Dusts | ||||
| Substance | Total | Respirable | Qualitative | |
| characterized | mg/m³ | mg/m³ | analyte(s) | |
|
| ||||
| Group I | ||||
| Aluminum | 15 | 5 | Al | |
| Bismuth telluride, undoped | 15 | 5 | Bi, Te | |
| Calcium carbonate | 15 | 5 | Ca | |
| Calcium silicate | 15 | 5 | Ca, Si | |
| Calcium sulfate | 15 | 5 | Ca, S | |
| Gypsum | 15 | 5 | Ca, S | |
| Limestone | 15 | 5 | Ca | |
| Marble | 15 | 5 | Ca | |
| Particulates not otherwise regulated | 15 | 5 | - | |
| Perlite | 15 | 5 | Si | |
| Plaster of Paris | 15 | 5 | Ca, S | |
| Group II | ||||
| Alpha-alumina | 10 | 5 | Al | |
| Ammonium sulfamate | 10 | 5 | S | |
| Emery | 10 | 5 | Al, Fe | |
| Kaolin | 10 | 5 | Al, Si | |
| Portland cement | 10 | 5 | Ca, Si | |
| Rouge | 10 | 5 | Fe | |
| Silicon | 10 | 5 | Si | |
| Silicon carbide | 10 | 5 | Si | |
| Group III | ||||
| Barium sulfate | 10 | 5 | Ba, S | |
| Dicyclopentadienyl iron | 10 | 5 | Fe | |
| Molybdenum, insoluble | 10 | Mo | ||
| Titanium dioxide | 10 | Ti | ||
| Zinc stearate | 10 | 5 | Zn | |
For all three groups listed, respirable dust samples are normally analyzed gravimetrically in the field. If crystalline silica is suspected, submit respirable samples to the lab for analysis.
Group I:
Sample analysis is based on a gravimetric determination performed in the field for total dust, because these PELs are the same as listed for "Particulates, not otherwise regulated". Additional analysis can be performed if necessary.
Group II:
Contact the laboratory before submitting samples, because methods may not be able to speciate the analyte.
4.2. Estimation of Aerosol Detection Limits (Back to Outline)
Experimental Design (Table 2a)
The detection limits for 21 elements were evaluated using aerosol air samples collected closed-face on tared PVC membranes. Element and reagent selection was based on the following considerations:
| a) | Elements found in dusts regulated by OSHA (Table 1) were included in order to provide estimates of detection limits for qualitative confirmations. |
| b) | Toxic elements which may be found while screening air samples were also included. If detected, samples containing these elements may be routed for appropriate analyses. |
| c) | Additional elements were selected to span the widest possible analytical range for each of the five secondary targets (Table 5b). In order to obtain estimates of the worst and best detection limits for each secondary target, analyses were performed on the least and most sensitive analytes. The analytical sensitivity for thin films is a smooth function of atomic number. This smooth function makes it possible to interpolate and extrapolate conservative detection limit estimates for elements not included in Table 2b (See Table 2c). |
| d) | When possible, realistic matrices were included. For
example, the National Institute of Standards and Technology
(NIST) Portland Cement Standard Reference Material #635
Also, lead chromate was considered a representative matrix for both Pb and Cr. |
| e) | Aerosol particles tend to concentrate in the center of air
filters when samples are collected using
|
The estimations of microgram detection limits for
| Table 2a Aerosol Analyte Detection Limit Determinations (Aerosol Source Materials) | |||||||
| Element | Source | Element | Source | Element | Source | ||
|
|
|
| |||||
| Al | AlPO4 | Cr | PbCrO4 | Ag | Ag | ||
| Si | SRM-635 | Mn | SRM-635 | Cd | Cd | ||
| P | AlPO4 | Fe | SRM-635 | Ce | Ce(OH)4 | ||
| S | SRM-635 | Zn | ZnO | Ho | Ho2O3 | ||
| K | SRM-635 | As | As2O3 | W | WO3 | ||
| Ca | SRM-635 | Sr | SrCO3 | Hg | HgO | ||
| Ti | TiO2, SRM-635 | Zr | ZrO2 | Pb | PbCrO4 | ||
| Table 2b Aerosol Analyte Detection Limit Determinations (Estimated Detection Limits) |
|||||||
| Element | kV Range | Micrograms | Analyte | Blank | Detection | Secondary | |
| from | to | Counts | Counts | Limit µ | Target | ||
|
|
|
|
|
|
|
| |
| Al Si P S K Ca Ti(TiO2) Ti(Blue) Cr Mn Fe Zn As Sr Zr Ag Cd Ce(La) Ho(La) W (La) Hg(La) Pb(La) |
1.330 1.540 1.800 2.100 3.120 3.420 4.280 4.360 5.180 5.740 6.140 8.380 10.360 13.840 15.360 21.680 22.640 4.600 6.440 8.120 9.620 10.160 |
1.640 1.920 2.250 2.600 3.460 3.890 4.730 4.650 5.650 6.070 6.650 8.880 10.700 14.440 16.120 22.480 23.560 5.050 7.000 8.660 10.320 10.900 |
194.0 176.6 222.8 58.1 7.7 877.4 140.3 3.9 222.4 1.3 37.5 46.6 6.8 38.6 299.8 36 201 202.6 644.3 508.3 1,041.9 886.0 |
967 1,070 4,817 3,440 2,066 173,044 5,517 156 14,631 164 7,418 2,184 84 932 9,226 612 2,783 3,768 36,032 6,213 47,456 28,014 |
1,673 3,999 8,017 115,906 193 248 41 31 69 57 62 25 15 20 20 159 158 41 36 58 38 36 |
24.62 31.31 12.42 17.25 0.16 0.24 0.52 0.42 0.38 0.18 0.12 0.32 0.94 0.56 0.44 2.22 2.72 10.3 0.32 1.87 0.41 0.57 |
Ti Ti Ti Ti Ti Ti Ge Ge Ge Ge Ge Zr Zr Ag Ag Gd Gd Ge Ge Zr Zr Zr |
Note: Membranes composed of PVC absorb low-energy
| Table
2c Conservative Estimated Aerosol Detection Limits (µg) | |||||||||||||||||
| PERIODIC TABLE | |||||||||||||||||
| H | He | ||||||||||||||||
| Li | Be | B | C | N | O | F | Ne | ||||||||||
| Na | Mg | Al 30 |
Si 30 |
P 20 |
S 20 |
Cl | Ar | ||||||||||
| K 2. |
Ca 2. |
Sc 1. |
[Ti] .5 |
V .5 |
Cr .4 |
Mn .2 |
Fe .1 |
Co .1 |
Ni .1 |
Cu .1 |
Zn 1. |
Ga 1. |
[Ge] 1. |
As 1. |
Se 1. |
Br 1. |
Kr |
| Rb 1. |
Sr .6 |
Y .5 |
[Zr] .4 |
Nb .4 |
Mo .4 |
Tc | Ru 6. |
Rh 5. |
Pd 4. |
[Ag] 3. |
Cd 3. |
In 3. |
Sn 3. |
Sb 3. |
Te 3. |
I 3. |
Xe |
| Cs 3. |
Ba 3. |
La 3. |
Hf 3. |
Ta 2. |
W 2. |
Re 2. |
Os 2. |
Ir 1. |
Pt 1. |
Au .8 |
Hg .6 |
Tl .6 |
Pb .6 |
Bi .5 |
Po | At | Rn |
| Fr | Ra | Ac | |||||||||||||||
| Between La and Hf: | |||||||||||||||||
| Ce 1. |
Pr 1. |
Nd 1. |
Pm | Sm .8 |
Eu .7 |
[Gd] .6 |
Tb .5 |
Dy .4 |
Ho .3 |
Er 4. |
Tm 4. |
Yb 3. |
Lu 3. | ||||
| After Ac: | |||||||||||||||||
| Th .5 |
Pa | U .5 |
Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lw | ||||
Microgram detection limits for elements in aerosols collected on
PVC are shown above. The detection limits are listed below the symbol
for each element that can be analyzed by this method. Results from
Table 2b were used to make conservative estimates for the 21 elements
evaluated (shown as bolded symbols). Detection limits for the
remaining elements that can be analyzed were next obtained by
interpolation and conservative extrapolation. All limits shown are
estimates. The noble gases, elements lighter than Al, and chlorine
cannot be analyzed on PVC membranes. (Chlorine and chlorine compounds
can be analyzed on MCE membranes.) Note: This method is not
appropriate for the radioactive elements Tc,
The secondary target elements used in this
method are enclosed in [ ].
Calculation of aerosol detection limits (Table 2b)
Detection limit calculations were performed as indicated in Section 1.3.1. of the method. [Note: Although widely used as an estimate of the qualitative detection limit, this theoretical approach assumes a model that does not consider effects from interferences. Also, special care was used when performing appropriate blank subtraction, background modeling, and profile fitting in order to isolate the light element fluorescence peaks.]
Discussion of aerosol detection limit results (Tables 2b-2c)
The analytical detection limits in Tables 2b-2c above were
determined using K analytical peaks, except as noted. Analyte counts
shown in Table 2b are rounded to the nearest whole count. With the
exception of the four lightest elements, the detection limits for most
of the elements are very low. Compared to loadings needed to
qualitatively analyze heavy elements on PVC membranes, relatively
large loadings are necessary for light elements. Because MCE membranes
are more transparent to
Additional recommendations for improving aerosol detection limits can be found in the Appendix.
4.3. Evaluation - Bulk Sample Determinations (Back to Outline)
Experimental design (Table 3 and Tables 4a-4d)
Recoveries for 37 elements in powdered heterogeneous and homogeneous bulk samples were evaluated in order to model typical samples that are sent to the laboratory. The following elements were incorporated in the study (listed in order of increasing atomic number):
Mg, Al, Si, P, S, K, Ca, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, As, Se, Br, Rb, Sr, Zr, Mo, Ag, Cd, In, Sb, Sn, Te, Ba, La, W, Hg, Tl, Pb, and Bi
The accuracy of this method is particularly sensitive to sample matrix effects, because standard matrices are not matched to sample matrices. For that reason, a wide variety of matrix types were used in the evaluation study. Homogeneous samples (Tables 3 and 4a) are useful in evaluating optimum conditions for analyses.
Heterogeneous samples (Tables 4b-4c) are useful in evaluating the effect of errors associated with packing and particle-size effects. They also have the additional error associated with obtaining representative samples of mixtures of solids. The evaluation samples consisted of seven known reference materials (Tables 4a-4b) and 12 evaluation bulk samples (Table 4c) prepared in a blind test of the method.
| a) | The results in Table 4a are for an organic (gelatin) matrix
containing trace elements in two standard reference materials
|
| b) | The results in Table 4b are for standard reference materials (SRMs) from NIST. These mineral samples were accompanied by certificates of analysis and represented intermediate weight element matrices. |
| c) | The results in Table 4c are for unknowns that were prepared in a manner to provide stable, challenging, and realistic samples of uniform composition. These mixtures were prepared by an independent chemist who ground and mixed the chemically compatible reagents. The majority of analytes were oxides. They included light, intermediate, and heavy matrices. |
The major component of each of the evaluation bulk samples in Table 4c was a matrix consisting of one or more of the following:
- boric acid (representing a light element matrix)
starch (representing a light element matrix)
zinc oxide (representing a heavy element matrix)
ferric oxide (representing a heavy element matrix)
silicon dioxide (Celite, representing an
Except for the ferric oxide, all the matrices were white; this reduced the analyst's ability to immediately assess the major components of each bulk. As the data began to accumulate, the analyst judged that the matrices could be arranged in groups of three. The analyst's observations during the blind experiments were:
| a) | Three samples (V4 through V6 listed below) tended to clump and gave strong signals for Zn. |
| b) | Three samples (V7 through V9) gave a strong signal for Si. The matrix identity of V1 through V3 and V10 through V12 could not be assessed from observations made by the analyst. |
The identities of the matrices were revealed after the results of the analyses were reported:
| Samples |
Matrix |
Type | ||
| V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 |
Boric acid Zinc oxide Silicon dioxide Corn Starch |
light element heavy element intermediate light element |
Prior to the analyses the analyst knew that oxides of the elements were the major materials used for the components. This information provided chemistry information during data workup. The analyst prepared an additional sample consisting of powdered aluminum oxide to check analytical sensitivity. Aluminum was the lightest element attempted in the analyses of samples V1 through V12.
Results were determined using three different software routines that streamline the following calculations:
The three approaches described in Sections 3.8.10.-12. of the
method were used to obtain quantitative estimates of the composition
of bulks presented in Tables
| a) | A custom procedure (results indicated by "QUANT" in Tables 3-4c) which calculated estimates only on detected elements. This procedure calls the proprietary Kevex fundamental parameters function QUANT/EXACT/FILM which takes into account the analytical data and the sample mass thickness. It performs an estimate of the composition of the sample in terms of analyzed elements (including any chemistry). |
| b) | A custom procedure (results indicated by "NORMQUANT" in Tables 3-4c) calls the proprietary Kevex function QUANT/EXACT/FILM/NORM which takes the result above and proportions the results so that the composition sums to 100%. |
| c) | A custom procedure (results indicated by "MARSQUANT" in Tables 3-4c) calls the proprietary Kevex software function QUANT/EXACT/FILM/MARS. Portions of the routine are iterative. It uses Compton and Rayleigh scatter data and MARS (described below) calibration data to correct for the presence of unanalyzed light elements. Warning messages are displayed when the scatter data are outside the calibration range or when the process does not converge (This occurs when the process fails to estimate a reasonable light element composition for the sample due to matrix effects). |
The MARS function accessed through the procedure "MARSQUANT" is a proprietary Kevex software function that is similar to the previously available Kevex CEMAS function. Portions of the routine are iterative. It appears to operate in the following sequence:
| 1) | The function QUANT/EXACT/FILM is called as described above producing an initial estimate of the sample composition in terms of analyzed elements (including optional chemistry). |
| 2) | The mean atomic number of analyzed elements (including optional chemistry, e.g. oxygen content in oxides) is next determined. |
| 3) | Using the calibration information and the Compton and Rayleigh scatter information, an estimate is made of the mean atomic number of all elements (analyzed and unanalyzed) in the sample. |
| 4) | The results from 2) and 3) above are used to estimate the mean atomic number of unanalyzed light elements (MZu). |
| 5) | Two light elements (E1 and E2) that bracket the mean atomic number of unanalyzed light elements are selected. The elements E1 and E2 need not be present in the actual sample; they are representative light elements used in computations only. |
| 6) | The corresponding atomic weights of these representative light elements, E1 and E2, are used to give a representative total weight fraction for the unanalyzed elements. |
| 7) | The remainder of the weight fraction is attributed to the analyzed fraction. |
| 8) | The analytical results of analyzed elements (including optional chemistry) from operation 1) are then scaled to equal the sum of the analyzed fraction obtained from operation 7). |
The overall composition includes the light elements that could be present in the sample. The analytical task was to determine the amount of each analyzed constituent relative to the overall composition of the sample. Test materials were analyzed using the three software routines listed above. For example, a test material consisting of a single analyzable constituent (e.g., Fe as Fe2O3) in a light element matrix might give disparate results consisting of:
For a single analyzable constituent, both QUANT and NORMQUANT always normalize to 100%; therefore, neither would be selected. If the MARS scatter data was within the calibration range, and MARSQUANT was able to converge, then the 3.1% result would be selected. If not, the analyst should consider reporting only qualitative results.
Results from only one of the three routines was selected for each
test material based on the criteria indicated below each of the
following tables of results. The reported results from that routine
were compared to the theoretical values for the test material. The
recovery for each analyzed element in each test material was
calculated. Statistics were evaluated for the recoveries for each test
material (where appropriate) and for all test materials. The recovery
data did not follow a normal distribution. A
The following characters, symbols, or nomenclature (in
RECOVERY = ratio of FOUND/THEORETICAL amounts
P = results in parts per million (µ/g)
1% = 10,000 µ/g
ND = None Detected
1SDƒ represents the factor used to
determine the
The low end of the recovery range for this analysis is obtained from:
- Mean recovery / 1SDƒ = 0.956 / 1.493 =
0.640
The high end of the recovery range is obtained from:
- Mean recovery × 1SDƒ = 0.956 × 1.493 =
1.427
Note: 2SDƒ = (1SDƒ)² 3SDƒ = (1SDƒ)³
Detected elements in the tables with recoveries in error by more
than a factor of 4 are flagged with the symbol "![]() ".
".
| Table 3 | ||||||||||
| SAMPLE (NH4)2SO4: | ||||||||||
| ELEMENT |
MARSQUANT% (REPORTED) |
QUANT% |
NORMQUANT% |
THEORETICAL% |
RECOVERY | |||||
| S | 34.49 | (100) | (100) | 24.27 | 1.421 | |||||
|
|
|
|||||||||
| Total | 34.49 | 24.27 | ||||||||
- QUANT and NORMQUANT both normalize to 100% when presented with
result files having only one analyzed component. MARSQUANT operated
without issuing error warnings and these results were selected. The
compound stoichiometry was not given to the software.
| SAMPLE AL2O3: | ||||||||||
| ELEMENT |
MARSQUANT% |
QUANT% |
NORMQUANT% (REPORTED) |
THEORETICAL% |
RECOVERY | |||||
| 0 Al Fe Zn Ga Zr |
26.77 30.08 197 P 24 P 124 P 50 P |
15.88 17.84 224 P 27 P 143 P 58 P |
47.04 52.83 665 P 81 P 424 P 173 P |
47.09 52.91 |
0.998 0.998 - - - - | |||||
|
|
|
|
| |||||||
| Total | 56.89 | 33.77 | 100.00 | 100.00 | ||||||
The MARS scatter corrections for light elements gave a mean atomic number (for all elements in sample) less than 0.5 Z above the highest MARS calibration standard (an arbitrary cut off at 11.15). However, no residual light elements were found by the MARS program. The results from either the QUANT or the NORMQUANT approaches better approximated this sample's composition; the sample was also known to be composed mainly of Al2O3. The NORMQUANT approach appeared most suitable in providing estimates of all constituents in the sample. This is representative of the utility of the method in estimating trace element composition when the major constituent is known and can be analyzed. This approach is used on some field samples, but it is not a strong test of the system.
| Table
4a Evaluation Bulk Sample Mixture Determinations Homogeneous Light Element (Gelatin) Matrix | ||||
| SAMPLE KODAK TEG50-B: | ||||
| ELEMENT | MARSQUANT % (REPORTED) |
THEORETICAL % | RECOVERY | |
| Na | 397 P | ND | ||
| Mg | 256 P | ND | ||
| Al | 60 P | ND | ||
| S | 0.52 | - | ||
| Cl | 0.91 | - | ||
| K | 88 P | - | ||
| Ca | 0.55 | 0.2025 | 2.716 | |
| Ti | 12 P | - | ||
| V | 6 P | - | ||
| Cr | 48 P | 47 P | 1.021 | |
| Mn | 49 P | 48 P | 1.021 | |
| Fe | 81 P | - | ||
| Co | 50 P | 46 P | 1.087 | |
| Ni | 46 P | 52 P | 0.885 | |
| Cu | 46 P | 51 P | 0.902 | |
| Zn | 41 P | 53 P | 0.774 | |
| As | 70 P | 115 P | 0.609 | |
| Se | 29 P | 39 P | 0.744 | |
| Ru | 12 P | - | ||
| Ag | 55 P | - | ||
| Cd | 42 P | 45 P | 0.933 | |
| Sb | 41 P | 57 P | 0.719 | |
| Te | 40 P | 45 P | 0.889 | |
| Ba | - | 50 P | ND | |
| Hg | 62 P | 55 P | 1.127 | |
| Tl | 56 P | 46 P | 1.217 | |
| Pb | 91 P | 59 P | 1.542 | |
| Bi | 22 P | 49 P | 0.449 | |
| MARS software ran without issuing error messages. Statistics for heavy certified elements (those beyond Ti) are shown as mean recovery data. | ||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.956, 1SDƒ = 1.493 | ||||
|
| ||||
| SAMPLE KODAK TEG50-C: | ||||
| ELEMENT | MARSQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| Li | 47 P | ND | ||
| Be | 42 P | ND | ||
| B | 51 P | ND | ||
| Na | 185 ±32 P | ND | ||
| Mg | 73 P | ND | ||
| S | 0.51 | - | ||
| Cl | 1.45 | - | ||
| K | 200 P | 94 ±32 P | 2.128 | |
| Ca | 1800 P | 570 ±53 P | 3.158 | |
| Ti | 13 P | - | ||
| P | 57 P | 52 P | 1.096 | |
| Cr | 54 P | 47 P | 1.149 | |
| Mn | 56 P | 45 P | 1.244 | |
| Fe | 72 P | 64 P | 1.125 | |
| Ni | 2 P | - | ||
| Cu | 60 P | 49 P | 1.224 | |
| Ga | 51 P | 48 P | 1.062 | |
| Rb | 39 P | 46 P | 0.848 | |
| Sr | 52 P | 48 P | 1.083 | |
| Zr | 48 P | 45 P | 1.067 | |
| Mo | 44 P | 59 P | 0.746 | |
| Ag | 111 P | 56 P | 1.982 | |
| In | 38 P | 48 P | 0.792 | |
| Sn | 37 P | 47 P | 0.787 | |
| Ba | 23 P | 44 P | 0.523 | |
| Bi | 46 P | 43 P | 1.070 | |
| MARS software ran without issuing error messages. Statistics for heavy certified elements (those beyond Ti) are shown as mean recovery data. | ||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.128 , 1SDƒ = 1.531 | ||||
| Table
4b Evaluation Bulk Sample Mixture Determinations Heterogeneous Intermediate Element (Mineral) Matrices | |||||
| SAMPLE SRM-635 (NIST Portland Cement "Blue"): | |||||
| ELEMENT | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | ||
| Volatiles | 3.24 | ND | |||
| B | <0.01 | - | |||
| F | 0.04 | ND | |||
| Na as Na2O | 0.07 | ND | |||
| Mg as MgO | 0.77 | 1.23 | 0.626 | ||
| Al as Al2O3 | 0.72 | 6.29 | 0.114 | ||
| Si as SiO2 | 7.28 | 18.4 | 0.396 | ||
| P as P2O5 | 0.17 | ND | |||
| S as SO3 | 8.15 | 7.07 | 1.153 | ||
| Cl | <0.01 | - | |||
| K as K2O | 1.17 | 0.45 | 2.600 | ||
| Ca as CaO | 80.08 | 59.83 | 1.338 | ||
| Ti as TiO2 | 0.17 | 0.32 | 0.531 | ||
| V | 19 P | <0.01 | - | ||
| Cr as Cr2O3 | 0.01 | 0.01 | 1.000 | ||
| Mn as MnA2O3 | 0.05 | 0.09 | 0.556 | ||
| Fe as Fe2O3 | 1.41 | 2.61 | 0.540 | ||
| Ni | 31 P | <0.01 | - | ||
| Cu | 12 P | <0.01 | - | ||
| Zn as ZnO | 0.01 | ND | |||
| Sr as SrO | 0.16 | 0.21 | 0.762 | ||
| Y | 14 P | - | |||
| Zr | <0.01 | - | |||
| Mo | <0.01 | - | |||
| Ag | <0.01 | - | |||
| Sn | <0.01 | - | |||
| Ba | 0.02 | <0.01 | - | ||
| Pb | <0.01 | - | |||
| This sample matrix was too heavy for successful MARSQUANT operation. Because the sample was known to be geological, normalized oxide results from NORMQUANT were selected as most representative. Sum peaks from strong Ca signals may be responsible for producing weak lines near Co and Ni analytical peaks (Small signals were noticed in the vicinity of the Co spectrum; however, results for Co were ND). The representation of analytes listed under the ELEMENT heading are as indicated in NIST certification documents. | |||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.677 , 1SDƒ = 2.217 | |||||
|
| |||||
| SAMPLE SRM-636 (NIST Portland Cement "Yellow"): | |||||
| ELEMENT | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | ||
| Volatiles | 1.16 | ND | |||
| B | <0.01 | - | |||
| F | 0.06 | ND | |||
| Na as Na2O | 0.11 | ND | |||
| Mg as MgO | 0.73 | 3.95 | 0.185 | ||
| Al as Al2O3 | 1.03 | 3.02 | 0.341 | ||
| Si as SiO2 | 9.88 | 23.22 | 0.425 | ||
| P as P2O5 | 0.08 | ND | |||
| S as SO3 | 2.33 | 2.31 | 1.009 | ||
| Cl | <0.01 | - | |||
| K as K2O | 1.38 | 0.59 | 2.339 | ||
| Ca as CaO | 83.48 | 63.54 | 1.314 | ||
| Ti as TiO2 | 0.11 | 0.18 | 0.611 | ||
| V | 0.01 | <0.01 | - | ||
| Cr as Cr2O3 | 0.00 | 0.01 | ND | ||
| Mn as Mn2O3 | 0.06 | 0.12 | 0.500 | ||
| Fe as Fe2O3 | 0.86 | 1.61 | 0.534 | ||
| Ni | 27 P | <0.01 | - | ||
| Cu | 31 P | <0.01 | - | ||
| Zn as ZnO | 0.01 | 0.03 | 0.333 | ||
| Rb | 9 P | - | |||
| Sr as SrO | 0.03 | 0.04 | 0.750 | ||
| Y | 15 P | - | |||
| Zr | 85 P | <0.01 | - | ||
| Mo | <0.01 | - | |||
| Ag | <0.01 | - | |||
| Sn | <0.01 | - | |||
| Ba | 0.06 | <0.01 | - | ||
| Pb | 0.01 | <0.01 | - | ||
| This sample matrix was too heavy for successful MARSQUANT operation. Because the sample was known to be geological, normalized oxide results from NORMQUANT were selected as most representative. Sum peaks from strong Ca signals may be responsible for producing weak lines near Co and Ni analytical peaks (Small signals were noticed in the vicinity of the Co spectrum; however, results for Co were ND). The representation of analytes listed under the ELEMENT heading are as indicated in NIST certification documents. | |||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.596 , 1SDƒ = 2.028 | |||||
|
| |||||
| SAMPLE SRM-637 (NIST Portland Cement "Pink"): | |||||
| ELEMENT | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | ||
| Volatiles | 1.69 | ND | |||
| B | <0.01 | - | |||
| F | 0.04 | ND | |||
| Na as Na2O | 0.15 | ND | |||
| Mg as MgO | 0.67 | ND | |||
| Al as Al2O3 | 3.28 | ND | |||
| Si as SiO2 | 9.04 | 23.07 | 0.392 | ||
| P as P2O5 | 0.24 | ND | |||
| S as SO3 | 1.72 | 2.38 | 0.723 | ||
| Cl | <0.01 | - | |||
| K as K2O | 0.97 | 0.25 | 3.880 | ||
| Ca as CaO | 87.00 | 66.04 | 1.317 | ||
| Ti as TiO2 | 0.09 | 0.21 | 0.429 | ||
| V | 0.01 | <0.01 | - | ||
| Cr as Cr2O3 | 0.01 | 0.01 | 1.000 | ||
| Mn as Mn2O3 | 0.04 | 0.06 | 0.667 | ||
| Fe as Fe2O3 | 0.94 | 1.80 | 0.522 | ||
| Co | 18 P | - | |||
| Ni | 38 P | <0.01 | - | ||
| Cu | 16 P | <0.01 | - | ||
| Zn as ZnO | 0.00 | 0.01 | ND | ||
| Sr as SrO | 0.07 | 0.09 | 0.778 | ||
| Y | 20 P | - | |||
| Zr | <0.01 | - | |||
| Mo | <0.01 | - | |||
| Ag | <0.01 | - | |||
| Sn | <0.01 | - | |||
| Ba | 0.10 | <0.01 | - | ||
| Pb | <0.01 | - | |||
| This sample matrix was too heavy for successful MARSQUANT operation. Because the sample was known to be geological, normalized oxide results from NORMQUANT were selected as most representative. Sum peaks from strong Ca signals may be responsible for producing weak lines near Co and Ni analytical peaks. The representation of analytes listed under the ELEMENT heading are as indicated in NIST certification documents. | |||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.820 , 1SDƒ = 2.012 | |||||
|
| |||||
| SAMPLE SRM-1881 (NIST Portland Cement "White"): | |||||
| ELEMENT | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | ||
| Volatiles | 2.01 | ND | |||
| B | <0.01 | - | |||
| F | 0.09 | - | |||
| Na as Na2O | 0.04 | - | |||
| Mg as MgO | 0.84 | 2.62 | 0.305 | ||
| Al as Al2O3 | 0.83 | 4.19 | 0.198 | ||
| Si as SiO2 | 9.83 | 22.25 | 0.442 | ||
| P as P2O5 | 0.09 | ND | |||
| S as SO3 | 4.57 | 3.65 | 1.252 | ||
| Cl | <0.01 | - | |||
| K as K2O | 2.23 | 1.17 | 1.906 | ||
| Ca as CaO | 78.71 | 58.68 | 1.341 | ||
| Ti as TiO2 | 0.12 | 0.23 | 0.522 | ||
| Cr | 28 P | <0.01 | - | ||
| Mn as Mn2O3 | 0.15 | 0.26 | 0.577 | ||
| Fe as Fe2O3 | 2.60 | 4.68 | 0.556 | ||
| Co | 68 P | - | |||
| Ni | 12 P | - | |||
| Cu | 12 P | - | |||
| Zn as ZnO | ND | 0.01 | ND | ||
| Rb | 4 P | - | |||
| Sr as SrO | 0.09 | 0.11 | 0.819 | ||
| Y | 11 P | - | |||
| Zr | 63 P | <0.01 | - | ||
| Ba | 83 P | <0.01 | - | ||
| This sample matrix was too heavy for successful MARSQUANT operation. Because the sample was known to be geological, normalized oxide results from NORMQUANT were selected as most representative. Sum peaks from strong Ca signals may be responsible for producing weak lines near Co and Ni analytical peaks. The representation of analytes listed under the ELEMENT heading are as indicated in NIST certification documents. | |||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.641 , 1SDƒ = 2.010 | |||||
|
| |||||
| SAMPLE SRM-2704 (NIST Buffalo River sediment): | ||||
| ELEMENT | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| Li | (50 P) | ND | ||
| C | 3.348 ±0.016 | ND | ||
| Na | 0.547 ±0.014 | ND | ||
| Al | 5.10 | 6.11 ±0.16 | 0.835 | |
| Si | 24.44 | 29.08 ±0.13 | 0.840 | |
| P | 0.998 ±0.0028 | ND | ||
| S | 1.08 | (0.4) | 2.700 | |
| Cl | (<0.01) | - | ||
| K | 7.36 | 2.00 ±0.04 | 3.68 | |
| Ca | 9.90 | 2.60 ±0.03 | 3.808 | |
| Sc | (12 P) | ND | ||
| Ti | 0.73 | 0.457 ±0.018 | 1.597 | |
| V | 144 P | 95 ±4 P | 1.516 | |
| Cr | 190 P | 135 ±5 P | 1.407 | |
| Mn | 963 P | 555 ±19 P | 1.735 | |
| Fe | 7.51 | 4.11 ±0.10 | 1.827 | |
| Co | 14.0 ±0.6 P | ND | ||
| Ni | 65 P | 44.1 ±3.0 P | 1.474 | |
| Cu | 206 P | 98.6 ±5.0 P | 2.09 | |
| Zn | 941 P | 438 ±12 P | 2.148 | |
| Ga | (15 P) | ND | ||
| As | 23.4 ±0.8 P | ND | ||
| Se | 4 P | (1.1 P) | 3.636 | |
| Br | 7 P | (7 P) | 1.000 | |
| Rb | 254 P | (100 P) | 2.540 | |
| Sr | 352 P | (130 P) | 2.708 | |
| Y | 79 P | - | - | |
| Zr | 797 P | (300 P) | 2.657 | |
| Nb | 35 P | - | - | |
| Cd | 3.45 ±0.22 P | ND | ||
| Sn | (9.5 P) | ND | ||
| Sb | 3.79 ±0.15 P | ND | ||
| I | (2 P) | ND | ||
| Cs | (6 P) | ND | ||
| Ba | 904 P | 414 ±12 P | 2.184 | |
| La | (29 P) | ND | ||
| Ce | (72 P) | ND | ||
| Sm | (6.7 P) | ND | ||
| Eu | (1.3) | ND | ||
| Dy | (6 P) | ND | ||
| Yb | (2.08 P) | ND | ||
| Lu | (0.6 P) | ND | ||
| Hf | (8 P) | ND | ||
| Hg | 53 P | 1.44 ±0.07 P | 36.8 | |
| Tl | 1.2 ±0.2 P | ND | ||
| Pb | 674 P | 161 ±17 P | 4.186 | |
| Th | (9.2 P) | ND | ||
| U | 3.13 ±0.13 P | ND | ||
| Noncertified theoretical values supplied by NIST are
shown in parentheses. This sample matrix was too heavy for successful MARSQUANT operation. The MARS approach failed on this geological material, so this material was analyzed as oxides and normalized to 100%. Error ranges are as indicated in NIST certification. | ||||
| Log-statistics (all detected analytes less
Hg): Mean recovery = 2.009 , 1SDƒ = 1.617 | ||||
| Log-statistics (all detected analytes including
Hg): Mean recovery = 2.308 , 1SDƒ = 2.201 | ||||
| Table
4c Evaluation Bulk Sample Determinations Heterogeneous Mixed Matrix Types (Blind Test) | ||||||
| SAMPLE V1: | ||||||
| ELEMENT | MARSQUANT
% (REPORTED) |
QUANT % | NORMQUANT % | THEORETICAL % | RECOVERY | |
| B | 11.61 | ND | ||||
| O | 7.81 | 14.37 | 40.05 | - | ||
| Na | 4.84 | ND | ||||
| Al | 1.37 | ND | ||||
| P | 0.96 | 1.05 | 2.92 | 3.26 | 0.294 | |
| K | 0.11 | 0.15 | 0.43 | - | ||
| V | 6.91 | 12.56 | 35.02 | 7.17 | 0.964 | |
| Fe | 1.82 | 5.79 | 16.13 | 1.42 | 1.282 | |
| W | 0.30 | 1.15 | 3.22 | 0.29 | 1.034 | |
| Hg | ||||||
|
|
|
| ||||
| Total | 18.14 | 35.87 | 100.00 | |||
| The MARS scatter corrections for light elements gave a mean atomic number (for all elements in sample) less than 0.5 Z above the highest MARS calibration standard (an arbitrary cut off at 11.15). Therefore, the results from the MARSQUANT approach were selected. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.612 , 1SDƒ = 2.216 | ||||||
|
| ||||||
| SAMPLE V2: | ||||||
| ELEMENT | MARSQUANT% (REPORTED) |
QUANT% | NORMQUANT% | THEORETICAL% | RECOVERY | |
| B | 15.80 | ND | ||||
| O | 1.89 | 13.94 | 28.02 | - | ||
| Al | 2.62 | ND | ||||
| P | 0.20 | 0.32 | 0.65 | - | ||
| S | 0.20 | 0.64 | 1.30 | - | ||
| Ca | 474 P | 0.24 | 0.48 | - | ||
| Cr | 0.75 | 4.27 | 8.58 | 0.98 | 0.765 | |
| Mn | 77 P | 450 P | 905 P | - | ||
| Fe | 346 P | 0.22 | 0.44 | 442 P | 0.783 | |
| Y | 162 P | 0.16 | 0.32 | - | ||
| Zr | 1.70 | 19.69 | 39.58 | 1.53 | 1.111 | |
| Mo | 0.49 | 5.93 | 11.92 | 0.45 | 1.089 | |
| Hf | 614 P | 0.41 | 0.83 | - | ||
| Pb | 0.52 | 3.87 | 7.79 | 0.46 | 1.130 | |
|
|
| |||||
| Total | 5.92 | 49.74 | 100.00 | |||
| No warnings were issued during the MARS scatter corrections for light elements. Therefore, results from the MARSQUANT approach were selected. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.961 , 1SDƒ = 1.219 | ||||||
|
| ||||||
| SAMPLE V3: | ||||||
| ELEMENT | MARSQUANT% (REPORTED) |
QUANT% | NORMQUANT% | THEORETICAL% | RECOVERY | |
| B | 15.20 | ND | ||||
| O | 2.11 | 9.39 | 27.58 | - | ||
| Al | 2.22 | ND | ||||
| V | 0.18 | 0.48 | 1.42 | 0.20 | 0.900 | |
| Cr | 85 P | 240 P | 705 P | - | ||
| Mn | 29 P | 110 P | 323 P | - | ||
| Fe | 4.05 | 17.00 | 49.90 | 4.34 | 0.933 | |
| As | 0.16 | 1.29 | 3.79 | 0.21 | 0.762 | |
| W | 0.42 | 2.93 | 8.68 | 0.37 | 1.135 | |
| Hg | 0.38 | 2.91 | 8.54 | 1.48 | 0.257 | |
|
|
|
| ||||
| Total | 7.31 | 34.07 | 100.00 | |||
| The MARS scatter corrections for light elements gave a mean atomic number (for all elements in sample) less than 0.5 Z above the highest MARS calibration standard (an arbitrary cut off at 11.15). Therefore, results from the MARSQUANT approach were selected. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.715 , 1SDƒ = 1.802 | ||||||
|
| ||||||
| SAMPLE V4: | ||||||
| ELEMENT | MARSQUANT% | QUANT% | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| O | 7.75 | 18.46 | 21.01 | - | ||
| Ti | 0.83 | 1.53 | 1.75 | 1.84 | 0.951 | |
| V | 0.10 | 0.19 | 0.22 | 0.22 | 1.000 | |
| Cr | 0.36 | 0.69 | 0.78 | 0.81 | 0.963 | |
| Mn | 33 P | 64 P | 73 P | - | ||
| Co | 29 P | 59 P | 67 P | - | ||
| Ni | 140 P | 286 P | 325 P | - | ||
| Cu | 116 P | 270 P | 3080 P | - | ||
| Zn | 26.16 | 63.49 | 72.25 | 73.39 | 0.984 | |
| Zr | 0.34 | 0.90 | 1.02 | 0.79 | 1.291 | |
| Mo | 0.81 | 2.13 | 2.42 | 1.66 | 1.458 | |
| Pb | 0.16 | 0.41 | 0.47 | 0.41 | 1.146 | |
|
|
|
| ||||
| Total | 36.54 | 87.86 | 100.00 | |||
| The MARS program extrapolated considerably beyond a mean atomic number = 11.15. The analyst decided that the results from either the QUANT or the NORMQUANT approaches adequately approximate this sample's composition. NORMQUANT was selected because the sample was known to be primarily oxides of the analyzed elements, and the MARSQUANT results suggested that the matrix did not have a large amount of unanalyzed light elements. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.100 , 1SDƒ = 1.182 |
||||||
|
| ||||||
| SAMPLE V5: | |||||||
| ELEMENT | MARSQUANT% | QUANT% | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | ||
| O | 6.88 | 17.21 | 19.90 | - | |||
| Al | 8.27 | - | |||||
| Cr | 730 P | 0.14 | 0.16 | 0.15 | (221 P) | 1.067 | |
| Mn | ( 36 P) | ||||||
| Fe | 483 P | 936 P | 0.11 | 93 P | (0.33) | 11.828 | |
| Ni | 146 P | 291 P | 337 P | (425 P) | - | ||
| Zn | 27.17 | 67.96 | 78.59 | 65.46 | (69.51) | 1.201 | |
| As | 0.20 | 0.54 | 0.63 | 0.70 | 0.900 | ||
| Zr | 0.18 | 0.50 | 0.58 | 0.37 | 1.568 | ||
| Mo | (578 P) | ||||||
| Cd | ( 86 P) | ||||||
| Hg | 1.05 | ND | |||||
|
|
|
| |||||
| Total | 34.58 | 86.47 | 100.00 | ||||
| The MARS program extrapolated considerably beyond a mean atomic number = 11.15. The analyst decided that the results from either the QUANT or the NORMQUANT approaches adequately approximate this sample's composition. NORMQUANT was selected because the sample was known to be primarily oxides of the analyzed elements, and the MARSQUANT results suggested that the matrix did not have a large amount of unanalyzed light elements. | |||||||
| This sample was digested using mineral acids and reanalyzed by ICP-AES. Results for those elements detected by the ICP analysis are shown in parentheses. | |||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.845 , 1SDƒ = 2.881 | |||||||
|
| |||||||
| SAMPLE V6: | ||||||
| ELEMENT | MARSQUANT% | QUANT% | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| O | 7.05 | 17.39 | 19.82 | - | ||
| Na | 4.37 | ND | ||||
| Si | 1.31 | ND | ||||
| P | 2.94 | ND | ||||
| Fe | 243 P | 417 P | 537 P | - | ||
| Ni | 174 P | 346 P | 394 P | - | ||
| Zn | 28.31 | 69.36 | 79.06 | 66.43 | 1.190 | |
| Br | 135 P | 355 P | 405 P | - | ||
| Zr | 0.13 | 0.33 | 0.38 | 0.25 | 1.520 | |
| Mo | 0.20 | 0.53 | 0.61 | 0.31 | 1.968 | |
| Hg | 0.19 | ND | ||||
|
|
|
| ||||
| Total | 35.78 | 87.74 | 100.00 | |||
| The MARS program extrapolated considerably beyond a mean atomic number = 11.15. The analyst decided that the results from either the QUANT or the NORMQUANT approaches adequately approximate this sample's composition. NORMQUANT was selected because the sample was known to be primarily oxides of the analyzed elements, and the MARSQUANT results suggested that the matrix did not have a large amount of unanalyzed light elements. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.527 , 1SDƒ = 1.286 |
||||||
|
| ||||||
| SAMPLE V7: | ||||||
| ELEMENT | MARSQUANT% | QUANT% | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| O | 36.57 | 35.16 | 46.70 | - | ||
| Al | 7.54 | ND | ||||
| Si | 28.18 | 26.08 | 34.65 | 31.34 | 1.106 | |
| K | 0.54 | 0.59 | 0.79 | - | ||
| Ca | 0.39 | 0.43 | 0.57 | - | ||
| Ti | 719 P | 793 P | 0.11 | - | ||
| Mn | 181 P | 214 P | 284 P | - | ||
| Fe | 8.08 | 9.69 | 12.87 | 9.24 | 1.393 | |
| Ni | 78 P | 104 P | 138 P | - | ||
| Cu | 21 P | 28 P | 37 P | - | ||
| Zn | 96 P | 127 P | 169 P | - | ||
| As | 0.70 | 0.94 | 1.25 | 1.88 | 0.665 | |
| Sr | 1.61 | 2.25 | 2.99 | 1.79 | 1.670 | |
| Ba | 113 P | 132 P | 176 P | - | ||
|
|
|
| ||||
| Total | 76.18 | 75.28 | 100.00 | |||
| The MARS program extrapolated considerably beyond a mean atomic number = 11.15 and residual light elements represented less than 50% of the sample. The analyst decided that the results from either the QUANT or the NORMQUANT approaches adequately approximate this sample's composition. NORMQUANT was selected because the sample was known to be primarily oxides of the analyzed elements, and the MARSQUANT results suggested that the matrix did not have a large amount of unanalyzed light elements. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.144 , 1SDƒ = 1.490 |
||||||
|
| ||||||
| SAMPLE V8: | ||||||
| ELEMENT | MARSQUANT% | QUANT% | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| O | 33.65 | 32.46 | 46.74 | - | ||
| Al | 3.61 | ND | ||||
| Si | 27.65 | 25.60 | 36.85 | 37.47 | 0.983 | |
| K | 0.61 | 0.78 | 1.12 | - | ||
| Ca | 0.48 | 0.62 | 0.90 | - | ||
| Ti | 626 P | 819 P | 0.12 | - | ||
| V | 41 P | 54 P | 78 P | - | ||
| Cr | 46 P | 61 P | 87 P | - | ||
| Mn | 24 P | 32 P | 46 P | - | ||
| Fe | 0.69 | 0.93 | 1.34 | - | ||
| Ni | 1.87 | 2.67 | 3.84 | 3.68 | 1.043 | |
| Zn | 144 P | 216 P | 312 P | - | ||
| Sr | 1.59 | 2.63 | 3.79 | 2.01 | 1.886 | |
| Zr | 37 P | 63 P | 91 P | - | ||
| Mo | 1.11 | 2.03 | 2.92 | 1.39 | 2.101 | |
| Ba | 47 P | 65 P | 93 P | - | ||
| W | 0.26 | 0.40 | 0.57 | 0.34 | 1.676 | |
| Hg | 0.32 | 0.49 | 0.71 | 1.61 | 0.441 | |
| Pb | 0.46 | 0.72 | 1.03 | 0.65 | 1.585 | |
|
|
|
| ||||
| Total | 68.79 | 69.45 | 100.00 | |||
| The MARS program extrapolated considerably beyond a mean atomic number = 11.15 and residual light elements represented less than 50% of the sample. The analyst decided that the results from either the QUANT or the NORMQUANT approaches adequately approximate this sample's composition. NORMQUANT was selected because the sample was known to be primarily oxides of the analyzed elements, and the MARSQUANT results suggested that the matrix did not have a large amount of unanalyzed light elements. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.442 , 1SDƒ = 1.816 |
||||||
|
| ||||||
| SAMPLE V9: | ||||||
| ELEMENT | MARSQUANT% | QUANT% | NORMQUANT% (REPORTED) |
THEORETICAL% | RECOVERY | |
| O | 38.54 | 37.40 | 47.03 | - | ||
| Na | 1.14 | ND | ||||
| Si | 31.03 | 29.28 | 36.82 | 36.02 | 1.022 | |
| P | 0.97 | ND | ||||
| K | 0.66 | 0.76 | 0.96 | - | ||
| Ca | 0.51 | 0.59 | 0.74 | - | ||
| Ti | 1.42 | 1.68 | 2.11 | 1.51 | 1.397 | |
| Cr | 0.43 | 0.52 | 0.65 | 0.53 | 1.226 | |
| Mn | 99 P | 122 P | 154 P | - | ||
| Fe | 0.70 | 0.87 | 1.09 | - | ||
| Ni | 1.18 | 1.50 | 1.88 | 1.98 | 0.949 | |
| Zn | 73 P | 94 P | 119 P | - | ||
| Rb | 25 P | 35 P | 44 P | - | ||
| Sr | 0.51 | 0.71 | 0.90 | 0.57 | 1.579 | |
| Zr | 2.02 | 2.89 | 3.63 | 2.34 | 1.551 | |
| La | 0.78 | 0.93 | 1.21 | 1.16 | 1.043 | |
| Hf | 416 P | 529 P | 665 P | - | ||
| Hg | 1.73 | 2.29 | 2.88 | 6.51 | 0.442 | |
|
|
|
| ||||
| Total | 79.56 | 79.53 | 100.00 | |||
| The MARS program extrapolated considerably beyond a mean atomic number = 11.15 and residual light elements represented less than 50% of the sample. The analyst decided that the results from either the QUANT or the NORMQUANT approaches adequately approximate this sample's composition. NORMQUANT was selected because the sample was known to be primarily oxides of the analyzed elements, and the MARSQUANT results suggested that the matrix did not have a large amount of unanalyzed light elements. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.082 , 1SDƒ = 1.507 | ||||||
|
| ||||||
| SAMPLE V10: | ||||||
| ELEMENT | MARSQUANT% (REPORTED) |
QUANT% | NORMQUANT% | THEORETICAL% | RECOVERY | |
| O | 0.51 | 5.43 | 17.10 | - | ||
| Mg | 0.23 | ND | ||||
| Al | 0.74 | ND | ||||
| Ti | 582 P | 0.30 | 0.94 | - | ||
| Cr | 0.44 | 3.26 | 10.27 | 0.51 | 0.863 | |
| Sr | 0.61 | 9.62 | 30.31 | 1.04 | 0.587 | |
| Ba | 0.42 | 5.35 | 16.87 | 0.56 | 0.750 | |
| La | 0.60 | 7.77 | 24.51 | 0.84 | 0.714 | |
|
|
|
| ||||
| Total | 2.63 | 31.72 | 100.00 | |||
| No warinings were issued during the MARS scatter corrections for light elements. Therefore, results from the MARSQUANT approach were selected. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.722 , 1SDƒ = 1.173 | ||||||
|
| ||||||
| SAMPLE V11: | ||||||
| ELEMENT | MARSQUANT% (REPORTED) |
QUANT% | NORMQUANT% | THEORETICAL% | RECOVERY | |
| O | 1.76 | 10.77 | 17.53 | - | ||
| Mg | 0.26 | ND | ||||
| Ti | 1.60 | 6.21 | 10.11 | 1.39 | 1.151 | |
| Fe | 474 P | 0.37 | 0.60 | 0.049 | 0.967 | |
| Sr | 0.44 | 3.97 | 6.47 | 0.36 | 1.222 | |
| Zr | 0.48 | 4.50 | 7.33 | 0.39 | 1.231 | |
| Ba | 3.59 | 35.44 | 57.70 | 4.16 | 0.863 | |
| Hf | 199 P | 0.16 | 0.26 | - | ||
|
|
|
| ||||
| Total | 7.94 | 61.42 | 100.00 | |||
| The MARS scatter corrections for light elements gave a mean atomic number (for all elements in sample) less than 0.5 Z above the highest MARS calibration standard (an arbitrary cut off at 11.15). Therefore, results from the MARSQUANT approach were selected. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 1.076 , 1SDƒ = 1.170 | ||||||
|
| ||||||
| SAMPLE V12: | ||||||
| ELEMENT | MARSQUANT% (REPORTED) |
QUANT% | NORMQUANT% | THEORETICAL% | RECOVERY | |
| O | 0.92 | 10.48 | 18.45 | - | ||
| Ti | 0.11 | 0.56 | 0.99 | - | ||
| Sr | 0.94 | 10.87 | 19.13 | 0.84 | 1.119 | |
| Zr | 1.27 | 15.50 | 27.29 | 1.11 | 1.144 | |
| Ba | 1.01 | 11.96 | 21.06 | 1.18 | 0.856 | |
| La | 0.61 | 7.06 | 12.42 | 0.81 | 0.753 | |
| Hf | 417 P | 0.37 | 0.66 | - | ||
|
|
|
| ||||
| Total | 4.89 | 56.80 | 100.00 | |||
| No warnings were issued during the MARS scatter corrections for light elements. Therefore, results from the MARSQUANT approach was selected. | ||||||
| Log-statistics (all detected analytes having
theoretical values): Mean recovery = 0.953 , 1SDƒ = 1.228 | ||||||
| Table
4d Worst-Case Bulk Detection Limits (µg/g) PERIODIC TABLE | |||||||||||||||||
| H |
He | ||||||||||||||||
| Li |
Be |
B |
C |
N |
O |
F |
Ne | ||||||||||
| Na |
Mg |
Al 8% |
Si 4% |
P 3% |
S 1% |
Cl .5% |
Ar | ||||||||||
| K 800 |
Ca 600 |
Sc 400 |
[Ti] 200 |
V 100 |
Cr 70 |
Mn 60 |
Fe 60 |
Co 50 |
Ni 50 |
Cu 50 |
Zn 50 |
Ga 50 |
[Ge] 50 |
As 120 |
Se 50 |
Br 50 |
Kr |
| Rb 50 |
Sr 50 |
Y 50 |
[Zr] 50 |
Nb 50 |
Mo 50 |
Tc |
Ru 50 |
Rh 50 |
Pd 50 |
[Ag] 50 |
Cd 50 |
In 50 |
Sn 100 |
Sb 60 |
Te 50 |
I 50 |
Xe |
| Cs 50 |
Ba 50 |
La 50 |
Hf 200 |
Ta 200 |
W 100 |
Re 100 |
Os 100 |
Ir 100 |
Pt 100 |
Au 100 |
Hg 1% |
Tl 50 |
Pb 100 |
Bi 50 |
Po |
At |
Rn |
| Fr |
Ra |
Ac | |||||||||||||||
| Between La and Hf: | |||||||||||||||||
| Ce 900 |
Pr 900 |
Nd 800 |
Pm |
Sm 700 |
Eu 700 |
[Gd] 600 |
Tb 600 |
Dy 500 |
Ho 500 |
Er 400 |
Tm 400 |
Yb 300 |
Lu 300 | ||||
| Ater Ac: | |||||||||||||||||
| Th 100 |
Pa |
U 100 |
Np |
Pu |
Am |
Cm |
Bk |
Cf |
Es |
Fm |
Md |
No |
Lw | ||||
- Bulk detection limits
Worst case percent detection limits for elements in bulk samples
are shown above. The detection limits are listed below the bolded
symbol for each element that was analyzed by this method. Results from
Tables
Discussion of bulk sample determinations Recovery results and outliers:
Generally, recoveries were excellent for pure compounds (Table 3)
and the trace elements in the gelatin standard reference materials
(Table 4a samples
a) The results had a large dynamic range.
b) Errors in this
analysis tend to accumulate not as the sum of many small errors, but
as the product of many small relative errors (factors differing
slightly from one).
A test of log-normality was performed. The standard deviation found in the log(RECOVERY) was 0.3028 corresponding to a 1SDƒ factor of 2.008 for recovery scatter. If ideal recovery at concentrations in the working range is taken as 1, a 1SDƒ factor of 2 has the following statistical consequences:
| ±SDƒ |
Recovery range |
Error range |
% of Samples (Frequency)* | ||||
| Factor |
Ideal = 1 |
Ideal = 0% |
Theory |
Found | |||
| 1SD ƒ = 2 | 1/2 to 2 | -50% to +100% | 68.3% | 76.1% | |||
| 2SD ƒ = 4 | 1/4 to 4 | -75% to +300% | 95.5% | 95.6% | |||
| 3SD ƒ = 8 | 1/8 to 8 | -88% to +700% | 99.7% | 98.1% | |||
* Frequency of samples, or area under the curve as designated by ±nSDƒ | |||||||
The following figure is a histogram describing the spread in recoveries for detected analytes having theoretical values:
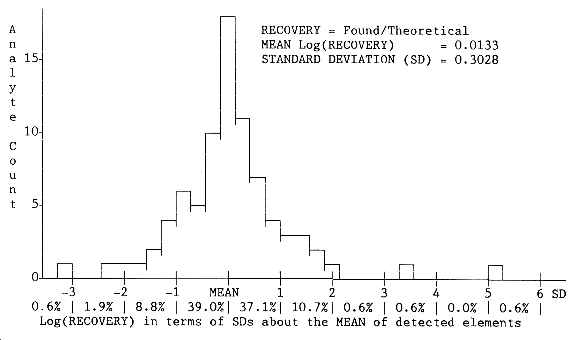
Figure 1
- The 1% DL for Hg shown in Table 4d is for a matrix containing ZnO; in the gelatin matrix Hg can be quantitated at 50 µg/g.
- The DL for As is large to compensate for the common interference from Pb.
- The DLs for Sn and Sb are large to compensate for an instrument artifact at the Sn Ka line.
- The DLs can change abruptly between elements using different secondary targets and line series. For the analysis of La, the Gd secondary target and Ka line are used; for the analysis of Ce, the Zr secondary target and La line are used.
- As examples:
Eighteen of the analytes were near the mean log(RECOVERY) of 0.0133, while one analyte was somewhat further than 3SD below this mean.
An attempt was made to estimate confidence limits for the results. The log(found µg/g) was fit as a linear function of the log(theoretical µg/g) shown in the following equation:
(i) (found µg/g) = 1.673 × (theoretical µg/g)0.9391
This equation indicates that results at low µg/g values (less than 4,767 µg/g) tend to err high, and higher µg/g results tend to err low.
The standard deviation in the calculated log(found µg/g) about the linear regression line obtained in this operation was approximately 0.2929. This corresponds to a 1SDƒ factor of 1.963 for the scatter in recovery comparable to 1SD (calculated from 100.2929). Because equation (i) has no significance other than described above and is not used to make any secondary corrections, the scatter associated with the following relation was evaluated:
(ii) (found µg/g) = 1.000 × (theoretical µg/g)1.000
The scatter of data about the line represented by equation (ii) was
evaluated by obtaining the standard deviation in the log (found µg/g)
about the individual log(theoretical g/g) values. The SD of
0.3023 corresponds to a 1SDƒ factor of 2.006
(calculated from 100.3023). When rounded to two significant figures,
both approaches give the same factor of 2.0 with the same statistical
consequences as noted in the table above. Two standard deviations is a
common criterion for determining outliers. In eight instances,
recoveries exceeded this criterion. These seven outliers (flagged with
the symbol "![]() " in the
tables) represent 4.4% of the 159 results used to evaluate recovery.
These results were not excluded when determining the statistics above.
The outliers included the two light elements Mg and Al and the three
heavy elements Fe, Hg, and Pb. Poor recoveries were noted also for K
and Ca in sample
" in the
tables) represent 4.4% of the 159 results used to evaluate recovery.
These results were not excluded when determining the statistics above.
The outliers included the two light elements Mg and Al and the three
heavy elements Fe, Hg, and Pb. Poor recoveries were noted also for K
and Ca in sample
The light elements Mg through K had poor recoveries in general
(samples
Results from ICP-AES support the higher level of Fe found by EDXRF (sample V5 in Table 4c). The mortar and pestle used in grinding and mixing the materials in Table 4c were unexpectedly difficult to clean. This outlier may be due to Fe contamination from this source.
The Hg outlier results were for sample SRM-2704 in Table 4b and
sample V1 in Table 4c. Analysis of sample
The Pb outlier result was also for sample SRM-2704 in Table 4b. This outlier overestimated by slightly more than the +2SD limit used to define an outlier. The presence of the unidentified trace element As was probably responsible.
In general, the recoveries were as expected for semiquantitative analysis for the elements heavier than calcium (excluding Hg as described above).
Detection limits are strongly influenced by matrix effects and instrumentation. The following are examples:
Non-detected elements heavier than Mg were present at levels exceeding 50 µg/g in some of the matrices. These included the following seven elements (Theoretical values are in parentheses):
Aluminum, Silicon, and Phosphorus:
Table 4c shows a large amount of undetected Al in sample V7 (7.54%) that is comparable to the amount found in V5 (8.27%) which was not known to contain any Al. The Al peak is a small shoulder on the strong Si peak in sample V7; Al is poorly resolved in this Si matrix. Once identified as present, the Al content changes from ND to 3.72%. Due to the low sensitivity for light elements, the small peak found in sample V5 near the Al spectrum calculates out to a large amount of Al due to the heavy (Zn) matrix.
The detection limits for Si and P are 4 and 3%, respectively, and the results for the two elements (V6 in Table 4c) are well below their detection limits for the matrices tested.
Chromium and Zinc:
Table 4b shows a Cr-containing sample (SRM-636) with Cr
non-detected. Table 4b also shows another Zn-containing sample
Cerium:
Table 4b shows a sample (SRM-2704) containing the
Mercury:
Mercury represents an exceptional heavy metal; several samples
contained Hg. While it performed well in the light matrix sample
If the seven outliers described above are excluded, the standard deviation in LOG(RECOVERY) was 0.2297. This corresponds to a 1SDƒ factor of 1.697 with the following statistical consequences:
| ±SDƒ |
Recovery range |
Error range |
% of Samples (Frequency)* | ||||
| Factor |
Ideal = 1 |
Ideal = 0% |
Theory |
Found | |||
| 1SDƒ = 1.697 | 0.589 to 1.697 | -41% to +70% | 68.3% | 69.5% | |||
| 2SDƒ = 2.880 | 0.347 to 2.880 | -65% to +188% | 95.5% | 94.0% | |||
| 3SDƒ = 4.888 | 0.205 to 4.888 | -80% to +389% | 99.7% | 100.0% | |||
* Frequency of samples, or area under the curve as designated by ±nSDƒ | |||||||
Non-certified Trace Element Composition:
Additionally, non-certified trace elements were detected in the bulk materials at levels exceeding 50 µg/g. These elements (listed in order of increasing atomic number) included the following:
Al, P, S, Cl, K, Ca, Ti, V, Cr, Mn, Fe, Ni, Cu, Zn, Ga, Br, Y, Zr, Ag, Ba, Hf
The trace elements in the reagents complicated the analyses. Some of the observations are mentioned:
a) Hf was present in the reagent used to provide Zr.
b) Trace contaminants were discovered in the Celite reagent. An EDXRF scan of the "pure" Celite material indicated that it had the following approximate composition:
| SiO2 | 94.84% | NiO | 0.03% | ||
| Fe2O3 | 2.18% | MnO2 | 0.02% | ||
| K2O | 1.35% | SrO | 0.01% | ||
| CaO | 1.25% | ZrO2 | 0.01% | ||
| TiO2 | 0.25% | CuO | (<0.01%> | ||
| V2O5 | 0.04% | Rb2O | (<0.01%> |
c) The pure Al2O3 (Table 3) contained detectable amounts of Fe, Zn, Ga, and Zr.
d) The ZnO was contaminated with several first transition elements.
e) The light matrices (boric acid and starch) are relatively free of trace contaminants.
4.4. Kevex Operating Conditions used in Evaluation (Back to Outline)
Experimental design (Table 5a-5b)
The conditions and data below are provided as suggested analytical
conditions for routine sample analyses using this method and to
describe overall instrument response. For
Kevex firmware and software use the term condition code
(abbreviated Cond. Code below). Each of the condition code numbers
Analytical preset times were all 200 s. Longer count times can be used if lower detection limits are necessary. The 12.5 µs time constant was used in order to obtain the best resolution for peak deconvolutions.
Reference elements (Ref) for fundamental parameters setup shown are
the pure sheet materials listed in Section 3.3. and analyzed at
the "Prescan mA" currents; the corresponding counts (Cts) are
integrated Gaussian peak areas for the
Also shown in the table are integrated Ka peak intensities in counts (at the "Prescan mA" setting) for the same reference materials that were used in the fundamental parameters (EXACT) calibration for the various condition codes. These reference materials were selected because they produced satisfactory quantitative estimates of bulk powder samples that were used in preliminary experiments. Other materials can be used.
| Table 5a | |||||||||
| Cond. Code |
kV |
Lucite mA |
Prescan mA |
Secondary Target |
Atmos. |
Range (kV) |
Preset Time |
Fund. Parameters
Ref Cts | |
| 1 | 60 | 0.830 | 0.130 | Gd | Air | 40 | 200 | Cu | 47,546 |
| 2 | 35 | 0.470 | 0.200 | Ag | Air | 40 | 200 | Cu | 315,627 |
| 3 | 25 | 2.360 | 0.550 | Zr | Air | 20 | 200 | Cu | 362,694 |
| 4 | 15 | 3.300 | 0.450 | Ge | Vacuum | 10 | 200 | Cu | 642,744 |
| 5 | 10 | 3.300 | 3.300 | Ti | Vacuum | 10 | 200 | Al | 28,900 |
Conditions for Air Samples:
Air samples typically give lower count rates than the Lucite monitor used in the analyses. The current settings in the "Lucite mA" column produce the maximum practical count rate for the Lucite monitor (not exceeding a 50% dead-time). The current was set to the "Lucite mA" values in order to produce the maximum feasible count rate in analyzing the filter samples.
Conditions for Bulk Samples:
Bulk samples can give count rates greater than that of the monitor.
The prescan currents in the column "Prescan mA" shown were selected so
that the majority of unknown bulk samples would give
The following is a rough guide to select the appropriate analytical ranges when deconvoluting elemental spectra for the different condition codes:
| Table 5b | ||||||
| Widest Element Ranges for Secondary Targets | ||||||
| Condition | Target | Ka | La | |||
| Code |
Element |
Range |
Range | |||
| 1 | Gd | Zr to La | --- | |||
| 2 | Ag | Cu to Rh | Hf to U | |||
| 3 | Zr | Cr to Rb | La to Bi | |||
| 4 | Ge | K to Cu | Sb to Ho | |||
| 5 | Ti | Al to Ca | Br to Sb | |||
| Optimum (non-overlapping) Element Ranges for Secondary Targets | ||||||
| Condition | Target | Ka | La | |||
| Code |
Element |
Range |
Range | |||
| 1 | Gd | Ru to La | --- | |||
| 2 | Ag | Sr to Tc | Tl to U | |||
| 3 | Zr | Zn to Rb | Ho to Hg | |||
| 4 | Ge | Sc to Cu | Sb to Dy | |||
| 5 | Ti | Al to Ca | Br to Sn | |||
4.5. Conclusions (Back to Outline)
Every attempt was made to mimic both air and bulk samples commonly received for qualitative analysis. The DLs and recoveries estimated for this method depend on how closely the samples resemble actual field samples. Analytical performance can be strongly influenced by interferences, sample matrix effects, and analyst experience.
For air samples, this method provides a useful tool to the industrial hygienist in confirming the presence of Table 1 substances in support of gravimetrically determined exposures. Other regulated elements may be identified in the process.
The method also provides a quick screen for up to 70 elements in
powdered bulk samples. Under certain circumstances it can also provide
quantitative estimates. Energy dispersive
5. References
- 5.1. Occupational Safety and Health Administration Analytical
Laboratory: Finnigan Standard Operating Procedure. Salt
Lake City, UT. 1979 (unpublished).
5.2. Occupational Safety and Health Administration Analytical
Laboratory: OSHA Analytical Methods Manual
5.3. Occupational Safety and Health Administration Technical
Center: Metal and Metalloid Particulates in Workplace Atmospheres
(ICP Analysis)
5.4. Birks, L.S.:
5.5. Bertin, E.P.: Principles and Practice of
5.6. Occupational Safety and Health Administration Analytical
Laboratory: Standard Operating Procedure, Inorganic Analysis by
(Back to Outline)
Additional recommendations to improve aerosol detection limits
Detection limits for some elements may be improved by redepositing
dust from the PVC sample medium onto
Longer integration times can also be used to reduce detection limits or to improve the precision in quantitation. The quality of analytical performance tends to be proportional to the square root of the analysis time.
Requests for the qualitative analysis of specific elements can
sometimes be given special attention; the instrument may be set up
with excitation conditions tailored to optimize for specific elements.
For example, an Fe secondary target may be used (instead of a Ge
secondary target) to give enhanced sensitivity for Cr. These
Additional recommendations to improve semiquantitative estimates
Semiquantitative XRF estimates can often be improved. Because XRF
analysis is
Additional improvements may be unnecessary in cases of