| Method Number: | ID-185 | |||||||||||||||
| Matrix: | Air | |||||||||||||||
Respirable dust: Fume: |
0.05 mg/m3 Time Weighted Average (TWA) 0.05 mg/m3 (TWA) | |||||||||||||||
Total dust: Fume: |
0.5 mg/m3 (Ceiling) 0.1 mg/m3 (Ceiling) | |||||||||||||||
| Sampler: | Low-ash polyvinyl chloride (PVC) membrane,
| |||||||||||||||
| Recommended Sampling Rate: | 1.7 liter per minute (L/min) | |||||||||||||||
| Analytical Procedure: | The PVC membrane is dissolved in tetrahydrofuran and a suspension is produced with the collected dust. The dust is transferred to a silver membrane for analysis by XRF and verification by XRD. | |||||||||||||||
| Recommended Air Volume: | 816 liters (8-h sample) For confirmation only | |||||||||||||||
(as V2O5) |
| |||||||||||||||
| Precision and Accuracy Validation Levels: |
0.29, 0.58, and 0.87 mg/m3 (as V2O5) using 237, 474, and 710 µg V2O5 loadings and assuming 816 liter air volumes | |||||||||||||||
Respirable Dust: CV1 Mean Recovery Overall Analytical Error |
| |||||||||||||||
| Method Classification: | Validated Confirmation Method | |||||||||||||||
| Chemist: | Mike C. Rose | |||||||||||||||
| Date: | October, 1987 (revised April, 1991) | |||||||||||||||
Similar products from other sources can b substituted.
OSHA Salt Lake Technical Center
Salt Lake City, Utah
1. Introduction
The goal of this method is to provide confirmation for occupational
vanadium pentoxide
(V2O5) exposures. To
achieve that end, the published
The procedure used in the NIOSH XRD study was adapted from the
published analytical procedure (8.1.)
and techniques (8.2.)
that were in print prior to the publication of NIOSH Method 7504 (8.3.).
The NIOSH sampling approach collects and analyzes only the respirable
fraction because of its toxic effects (8.1.).
[Vanadium pentoxide is also toxic by other routes of exposure
Particle size effects on the analysis were investigated during the OSHA
validation when the OSHA PEL was for total dust and fume (i.e.
Transitional PELs). The respirable
- 1.1. History
The previous two OSHA methods for V were atomic absorption
spectroscopy (AAS) (8.8.)
and inductively coupled plasma atomic emission spectroscopy
This method was validated in 1987 using a Finnigan
1.2. Analytical Principles
- 1.2.1. XRD:
Quantitative powder XRD relies upon the diffraction of
monochromatic
n(l) = 2d sin (q)
1.2.2. XRF:
Thin film quantitative
1.2.3. XRD and XRF:
For uniform thin films, these
1.3. Advantages and Disadvantages
- 1.3.1. Advantages of both methods:
Both methods are
1.3.2. Advantages of XRD:
The XRD method is specific for determining V2O5. The sampling and analytical approach provides the opportunity for concomitant analysis for respirable quartz and/or zinc oxide by XRD (8.11. and 8.12.).
1.3.3. Advantages of XRF:
Energy dispersive XRF provides the opportunity for the serendipitous discovery of other toxic elements that might be present in the air sample. For example, Pb and U are present in many V ores (8.1., 8.6.).
Detection limits may be significantly decreased for the XRF analysis by increasing the data collection time.
1.3.4. Disadvantages of both:
The XRF and XRD instrumentation are expensive.
1.3.5. Disadvantages of XRD:
Quantitation by XRD is
The presence of a
Preferred-orientation effects are potentially large for XRD analyses of V2O5, due to cleavage along favorable planes (8.1.).
1.3.6. Disadvantages of XRF:
X-ray fluorescence (XRF) cannot speciate the
1.4. Vanadium Pentoxide (CAS
| Application
|
Source of Exposure
| |
| catalyst | oxidation of nitrogen and sulfuroxides | |
| colorant | manufacture of yellow glass | |
| developer | photography industry | |
| coating | using welding rods | |
| alloys | manufacture of special steels | |
| contaminant | cleaning fuel oil burners |
1.5. Physical and Chemical Properties (8.5., 8.6.):
| Molecular weight: | 181.90 |
| Specific gravity: | 3.35 |
| Melting point: | 690 °C |
| Boiling point: | 1750 °C (decomposes) |
| Vapor pressure: | non-volatile at room temp. |
| Aqueous solublility: | 0.8 g/100 mL at 20 °C |
1.6. Toxicology
Information contained in this section is a synopsis of current knowledge of the physiological effects of V2O5 and is not intended as a basis for OSHA policy.
- 1.6.1. When inhaled, the chief effects of
V2O5 are on the
respiratory passages. Tracheitis, bronchitis, emphysema, pulmonary
edema, or bronchial pneumonia may be observed, but no specific
chronic lung lesions have been described. Other symptoms reported
include eye irritation, conjunctivitis, dermatitis, green tongue,
metallic taste, throat irritation, increased mucus, and cough (8.6.).
1.6.2. The toxic effects of
V2O5 are
primarily from exposures to dusts in the respirable
1.6.3. Death has been observed when animals were exposed to 70 mg/m3 for a few hours (8.6.).
2. Range, Detection Limit, and Sensitivity (8.10.)
This method was evaluated when the OSHA PELs were Ceiling total dust
and fume values. It should perform well under the more recent
- 2.1. Loadings of 237 to 710 µg
V2O5 were
evaluated. The calibration curves are usable up to at least 2,500 µg
V2O5, the highest
loading used in the method. Both
2.2. The quantitative detection limits (DL) for
V2O5 depend upon
the
| X-ray Fluorescence
| |||||||
| X-ray Diffraction
|
Direct-Beam
|
Secondary Target
| |||||
| DL
|
Total time
|
DL
|
Total time
|
DL
|
Total time
| ||
| 25 µg 20 µg |
65 s 650 s |
2 µg |
100 s 1,000 s |
0.25 µg 0.10 µg |
100 s 600 s | ||
2.3. The sensitivity (S) expressed in counts/µg (as V2O5) is also dependent upon analytical time:
| X-ray Fluorescence
| |||||||
| X-ray Diffraction
|
Direct-Beam
|
Secondary Target
| |||||
| S
|
Total time
|
S
|
Total time
|
S
|
Total time
| ||
| 18 180 |
65 s 650 s |
4 40 |
100 s 1,000 s |
120 720 |
100 s 600 s | ||
3. Method Performance (8.10.)
- 3.1. X-ray Instrumentation
The XRF portion of the method was evaluated in 1987 using a
In order to assess the method performance as a function of particle
size, both coarse and fine
V2O5 respirable
dust materials were investigated during the evaluation of this method.
Both materials passed through a
| Material
Coarse Fine |
0.5 to 3 µm
50% 99% |
3 to 10 µm
50% 1% |
These results are approximate and are only used to show that the median visible particle size of the coarse material approximated the 3.5 µm median diameter that is characteristic of respirable dust (8.13.). This coarse material was referred to as "respirable dust" in the evaluation and was used to assess the method performance for respirable dust.
Particles below 0.5 µm could not be counted directly using optical
microscopy. Visual observations of acetonitrile suspensions of the
coarse and fine materials indicated that a significant amount of the
fine material had a very small particle size; after thorough mixing,
the suspension of the coarse material cleared over the course of a day
whereas the suspension of the fine material remained cloudy for
several days. The fine material consisted of particles of 3 µm or less
and had a
| Dust
|
X-ray Diffraction
|
X-ray Fluorescence
| |
| Respirable | CV1 Range | 0.0555 - 0.0830 | 0.0392 - 0.0795 |
| Respirable | CV1 Pooled | 0.068* | 0.064 |
| Fine-respirable | CV1 Range | 0.0946 - 0.1663 | 0.0612 - 0.1456 |
| Fine-respirable | CV1 Pooled | 0.121* | 0.097 |
| Respirable | Recovery Range | 163.4 - 190.2% | 95.7 - 97.6% |
| Respirable | Recovery Mean | 177.4%* | 96.5% |
| Fine-respirable | Recovery Range | 85.9 - 91.1% | 84.8 - 89.4% |
| Fine-respirable | Recovery Mean | 88.7%* | 87.1% |
| Respirable | Overall Error | ± 91% | ± 16% |
| Fine-respirable | Overall Error | ± 36% | ± 32% |
* Average results from two APDs sharing the same generator.
These results indicate there are unacceptably severe errors
associated with
After the method validation, a
- 4.1. XRD
For XRD, any crystalline material with a diffraction peak in the
location of the XRD diffraction line at 31.05° 2q is an interference. An earlier study (8.1.)
indicated no interferences in common mineral compositions that contain
V2O5. During the
evaluation (8.10.),
a significant
Note: The potential interferences for XRF were assessed using a Link Systems EDX slide rule (Nashua Corp., Los Angeles, CA) to gauge other analyte signals and their proximity to the V Ka and V Kb peaks.
The V XRF lines have potential positive interferences. Known potential interferences are tabulated in Table 1. Depending on the resolution of the instrument and the available deconvolution software, the most significant likely interferences on an instrument with a resolution of 0.15 to 0.17 kV peak width at half height are the Ti Kb peak which overlap the V Ka peak and the Cr Ka peak that occur at the V Kb peak. Depending on the instrument geometry, Ag diffraction peaks from the Ag membrane are also possible. The Ti Ka peak may interfere with background correction.
Sum peaks occur when the counting rate is too high to accurately
measure the energies of the
When present in large amounts, iron and cobalt could give Si escape
peaks in the vicinity of the V Ka and Kb positions. An escape peak is
generated by the
Absorption edges (ab), are step discontinuities in the Energy vs.
The remaining interferences are the L lines of other elements which, when present, tend to be of low intensity.
5. Sampling
- 5.1. Sampling Equipment
- 5.1.1. Sample assembly:
Filter holder consisting of a
Backup pad,
Low ash PVC
membrane filter,
Note: During preparation for analysis, the sample is dissolved in tetrahydrofuran (THF). Certain acrylic copolymers added to PVC filters are insoluble in THF. If the membrane filter composition is unknown, a laboratory test should be conducted with THF to determine suitability before use.
5.1.2. Cyclone: Nylon,
Note: A cyclone is optional for fume sampling. If a mixture of fume and dust is anticipated, a cyclone should be used.
5.1.3. Pump calibration system: Stop watch and bubble tube or electronic meter.
5.1.4. Sampling pump: Calibrate the personal sampling pump to
approximately 1.7 L/min. Use a representative sampler (cyclone,
filter, etc.)
5.1.5. Assorted flexible tubing.
5.1.6. High volume sampling pump with cyclone (optional - for
bulk sample collection). Whenever possible, take a high volume
respirable V2O5
dust sample for laboratory use as a respirable dust reference
material. This is beneficial because
V2O5 has
significant
5.2. Sampling Procedure
- 5.2.1. Place the PVC filter and a cellulose backup pad in a two
or three piece cassette.
5.2.2. Attach the cassette, which is preceded by a
5.2.3. Place the sampling assembly in the breathing zone of the worker or sampling area and place the pump in an appropriate position. Take 816 L of air through the cassette at approximately 1.7 L/min. Do not allow the cyclone to be inverted during or after sampling. Take full shift samples if possible. Collect fume samples without a cyclone at 2 L/min flow rates.
5.2.4. Check the pump and sampling assembly periodically to verify performance and to monitor particulate loading on the sample filter. If the filter becomes overloaded (>3 mg) during the sampling interval, replace it with another filter.
5.2.5. Terminate sampling at the predetermined time and record the pump flow rate and collection time.
5.2.6. Record on the OSHA 91 form all pertinent sample data. When other compounds are known or suspected to be present in the air, such information, including their suspected identities, should be transmitted with the samples. Request "confirmation analysis of vanadium pentoxide".
5.2.7. Identify and submit an appropriate blank filter from each lot of filters used.
5.2.8. Seal each filter cassette and identify it with an OSHA Form 21. Mail samples to the laboratory in a suitable container designed to prevent damage.
5.3. Wipe samples are not appropriate for this analysis, but may be submitted for analysis by ICP-AES.
5.4. Bulk samples are appropriate, especially if they represent settled dust.
5.5. Bulk samples should be shipped separately from air samples. They should be accompanied by Material Safety Data Sheets if available. A description of the sample composition is useful in resolving interferences and should accompany bulk samples. Check current shipping restrictions and ship to the laboratory by the appropriate method.
6. Analysis
Samples are analyzed by both XRD and XRF. Only one sample preparation method is necessary; both analytical techniques are compatible with this sample preparation.
- Indium-lined collimators.
- Polypropylene window film, 0.20 mil thick (part no. 3520, SPEX Industries, Edison, NJ).
- Rhodium
X-ray tube. - Laboratory press, 12 ton (Cat. no. A14-100, Kevex, San Carlos, CA.).
- Lithium-drifted silicon (SiLi) detector.
- Radiation safety monitor (S.E. International Instrumentation
Model Radiation Alert Monitor 4, S.E. International
Instrumentation Division, Summertown, TN). This safety monitor can
be used with other
X-ray equipment below. - Lucite monitor.
- Tantalum collimator.
- Iron secondary target.
- Sample holders for
25-mm diameter Ag membranes (Cat. no.A00-213, Kevex). Note: These holders may require light machining in order to center the Ag membrane over the most sensitive Spot. - Pellet die set for preparing
multi-channel analyzer (MCA) energy calibration sample,31-mm diameter (Cat. no.A10-403, Kevex.) Alternately, a13-mm diameter die set (Cat. no.A10-401, Kevex) may be used. - Sample holder for
31-mm diameter MCA energy calibration sample (Cat. no.A00-214, Kevex). Note: If a13-mm diameter pellet is used, substitute a13-mm diameter sample holder (Cat. no.A00-212, Kevex). - Automated Powder Diffractometer (APD).
X-ray Generator.- Long,
fine-focus copper targetX-ray tube. - Scintillation, position sensitive, or xenon proportional counter detector.
X-ray accessories include:pulse-height analyzer, graphite monochromator, 2q compensating slit, 1° receiving slit, and sample spinner.- Sample holders for
25-mm diameter Ag membranes (Model no. PU1813/26, Philips Electronics Instruments Co., Mahwah, NJ). - Recirculating cooling system for the
X-ray tube. - Hardware and software for data reduction and graphic presentations.
- Line profile library
(JCPDS-International Center for Diffraction Data Powder Diffraction File, JCPDS, Swarthmore, PA). - Low ash PVC membrane filters,
37-mm, 5-µm pore size [part no. 625413, Mine Safety Appliances (MSA), Pittsburgh, PA or cat. no.P-503700, Omega Specialty Instrument Co., Chelmsford, MA]. - Analytical balance capable of 10 µg precision.
- Centrifuge tubes: Round bottom
40-mL (Pyrex 8260). - Gloves, THF-resistant [such as latex gloves (Cat. no. 8852, American Pharmaseal Lab., Glendale, CA)].
- Volumetric pipettes, eyedropper, volumetric flasks and graduated cylinders.
- Magnetic stirring bar and stirrer.
- Forceps.
- Silver membrane filters:
25-mm diameter,0.45-µm pore size (Cat. no.FM25-0.45, Osmonics, Inc., Minnetonka, MN). - Ultrasonic bath.
- Filtration apparatus:
25-mm (Filter Holder Hydrosol Manifold, cat. no.XX25 047 00, filtering clamps, cat. no.XX10 025 03, fritted glass bases with stoppers, cat. no. XX10 025 02, and glass funnels, cat. no.XX10 025 11, Millipore Corp., Bedford, HA). - Liquid nitrogen
cold-trap system for THF collection (dewar, polypropylene2-liter suction flask, liquid nitrogen, etc.). - Eyedropper.
- Hot plate, intrinsically safe (Model HP-11515B, Sybron/Thermolyne, Dubuque, IA).
- Teflon sheet, 0.3 to 1 mm thick (cut to fit top of hot plate).
- Plastic petri dishes (Product no. 7242, Gelman Sciences, Ann Arbor, MI).
- Vacuum system.
- Freezer mill (Model no. 6700, Spex Industries, Edison, NJ).
- Sieve, nylon, 10 µm (Spectra/Mesh N sieve, Cat. no.
08-670-205, Fisher Scientific, Springfield, NJ) or (Cat. no. 146514, Spectrum Medical Industries, Inc., Los Angeles, CA). - Polypropylene
250-mL wash bottle with tube cut out of top. - Mortar and pestle.
- Sieve or sonic sifter: Sieve,
325-mesh, (or Model ATML3P Sonic Sifter with325-mesh sieve, ATM Corporation, Milwaukee, WI). - Tetrahydrofuran
- Vanadium pentoxide
- Liquid nitrogen (for vapor trap and for maintaining EDXRF SiLi detector).
- Titanium dioxide
- Zinc oxide
- Yttrium oxide
- Boric acid (for
secondary-target instrument) - Bulk samples approximating respirable particle size:
Weigh an aliquot of 1 to 2 mg on a PVC filter, and place in a round bottom centrifuge tube. Prepare as for air samples using the procedure in Section 6.5.2., steps3-8. - Non-respirable bulks:
Grind the sample to a fine powder using either a mortar and pestle or a freezer mill). Then size the sample, using a325-mesh sieve or sonic sifter. This results in a sample particle size of less than 45 µm. Weigh out 1 to 2 mg of the sized sample on a PVC filter and place it in a round bottom centrifuge tube. Prepare as for air samples using the procedure in Section 6.5.2., steps3-8. - When sample weights are greater than 2.5 mg, aliquots are taken to achieve depositions within the working range.
- Examine the filter and backup pad to determine if any breakthrough to the backup pad has occurred. If there is significant breakthrough, the sample is either not analyzed or results are reported with a disclaimer. (See Section 7.3. for reporting results.)
- Carefully transfer the air sample (PVC filter) from the
cassette to
around-bottom 40-mL centrifuge tube. Add 10 mL THF to dissolve the filter and suspend the sample. Sonicate the sample suspension for 5 to 10 min. - For each sample, center a Ag membrane on the
fritted-glass base of the filtering apparatus. Also center the glass chimney on top of the base and secure it with a clamp. - With the vacuum turned off, place 2 to 5 mL of THF in the chimney of the previously assembled vacuum filtering apparatus.
- Quantitatively transfer the suspension with rinses of THF to the glass chimney of the vacuum filtering apparatus. The total volume in the chimney should not exceed 20 mL.
- Continue preparation of samples as described Sections 6.4.12. to 6.4.15.
- Consult the XRF SOP (8.16.)
and power up the
X-ray tube to 20 kV and 0.5 mA. Remove any filter and put the narrow collimator in place over the detector. Calibrate the element markers using aTi-Zn-Y calibration standard consisting of a1- to3-mm deep equimolar mix of powdered TiO2, ZnO, and Y2O3 on a 0.25 mil mylar support. Calibrate on the corresponding Ka lines at 4.508, 8.631, and 14.933 kV. - First analyze the standards. Next analyze the samples and
repeat the standards. For each sample or standard perform the
following steps:
- Close the shutter and open the cover.
- Wipe the yellow Kapton window before analyzing each sample.
- Place the sample face down on the yellow Kapton window over the detector, close the cover, open the shutter and initiate analysis.
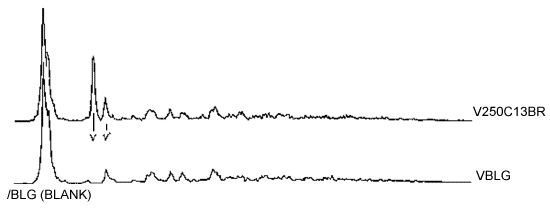
- Label the data to identify standards and samples analyzed. Typical spectra for a 250 µg V2O5 standard and a blank are shown in Figure 2.
- Save each spectrum to magnetic tape or disk (if possible). Note: On certain XRF systems without mass storage, it may be necessary to perform all the calculations after each sample is analyzed. Others allow some calculations to be incorporated automatically in the analyses prior to saving the spectra.
- Open sample chamber lid.
- Install a Ta collimator at the detector if not already installed.
- Install the Fe secondary target if not already installed.
- Place Ag membranes in the autosampler. Position each sample for maximum analytical signal during analysis.
- Close sample chamber lid.
- Calibrate the MCA using the
Ti-Zn-Y calibration standard consisting of a pellet made of a mixture H3BO3 and10-20% of an equimolar mix of powdered TiO2, ZnO, and Y2O3. Calibrate on the corresponding Zn and Y Ka lines at 8.631 and 14.933 kV respectively using a Ag secondary target. One may substitute other appropriate calibration materials or secondary targets for this purpose. - Select the following conditions:
Fe secondary target.
Display range = 10 kV.
X-ray tube power = 12 kV and 3.3 mA.
(To reduce the affect of sum peaks, do not exceed 50% deadtime on standards or samples.)
Atmosphere = Vacuum.
Acquisition time = 600 s.
Analyze a Lucite monitor sample or other appropriate monitor sample to adjust for varyingX-ray tube output. - Analyze all standards and samples.
- Save spectra to magnetic tape or disk. Note: On certain XRF systems it may be convenient to perform escape and sum peak corrections operations prior to saving each spectrum.
- 6.1. Safety Precautions
- 6.1.1. Follow laboratory safety rules and regulations regarding
solution preparation and instrument operation.
6.1.2. Refer to the appropriate manuals for proper instrument
operation and maintenance. Analysts that are unfamiliar with the
instrumentation must be trained prior to using the equipment. Refer
all
6.1.3. Tetrahydrofuran (THF) is extremely flammable and tends to form explosive peroxides. If static electric discharge is a potential problem, use a grounded wrist strap when transferring THF. Fires involving THF are likely to rapidly become fierce; in case of a large fire, sound an alarm and evacuate.
6.1.4. Use appropriate caution when handling chemicals. Use latex
or other
6.2. Equipment
Either a
- 6.2.1. Direct-beam XRF:
The spectrometer should be equipped with appropriate monitors and
collimators. The spectrometer used at the OSHA Salt Lake Technical
Center
6.2.2. Secondary-target XRF:
The spectrometer should be equipped with appropriate monitors,
collimators, and secondary targets. In addition to the accessories
listed in 6.2.1. items
6.2.3. XRD system consisting of:
6.2.4. Computer system consisting of:
6.2.5. Standard and sample preparation:
6.2.6. Bulk sample preparation for membrane deposition:
6.3. Reagents (except for liquid nitrogen use reagent grade or better):
- 6.3.1. Sample and standards preparation
6.3.2. Reagents for preparing MCA calibration sample
6.4. Preparation of Standard Materials
- 6.4.1. Use reagent grade
V2O5 as the
starting material.
6.4.2. Prepare respirable stock material by grinding reagent
grade V2O5 in a
6.4.3. Add this ground material to 50 to 75 mL of THF in a
6.4.4. Remove the tube from the cap of a
WET-SIEVE METHOD FOR STANDARD PREPARATION

6.4.5. Isolate the dust from the suspension by removing the suspension from the bottle. Filter the dust onto a Ag membrane. A filtering apparatus similar to the one described below can be used.
6.4.6. Allow the residual THF on the dust to evaporate at room
temperature. Remove the dust from the Ag membrane and store this
respirable stock material in a
6.4.7. Assemble the filtering apparatus and liquid nitrogen cold trap, and connect to a vacuum pump. Connect the cold trap in between the filtering apparatus and vacuum pump to collect the waste THF. Any waste vapors should not enter the vacuum pump.
6.4.8. Prepare a 10 µg/mL
THF-V2O5
suspension by weighing out 5 mg of respirable
V2O5 stock
material and quantitatively transferring it to a
6.4.9. For each standard, center a Ag membrane on the
6.4.10. With the vacuum turned off, place 2 to 5 mL of THF in the chimney of the previously assembled vacuum filtering apparatus.
6.4.11. Turn off the stirrer and shake the suspension vigorously prior to removing each aliquot from the center of the THF suspension. Consult the following table for determining the appropriate reagent concentration and aliquot size to take in preparing each of 24 calibration standards (three standards at each of the eight levels):
| Standard µg V2O5 |
Volume (mL) |
Reagent Concn (µg/mL) | |||
| 50 100 200 250 500 1,000 2,000 2,500 |
5 10 2 25 5 10 20 25 |
10 10 100 10 100 100 100 100 |
|||
Note: Duplicate standards are prepared so that outliers may be identified and discarded. Subsequent calibrations can be performed using fewer standards at levels selected to be comparable to the samples.
6.4.13. Carefully disassemble the chimney and clamp. Remove the
Ag membrane from the
6.4.14. When dry, place the standard in a labeled petri dish.
6.4.15. Place any THF waste in an
6.5. Preparation of Bulk and Air Samples
- 6.5.1. Bulk sample preparation:
To prevent the possibility of contaminating air samples, prepare
bulk samples in an area away from where air samples are prepared; a
separate filtering apparatus is recommended.
6.5.2. Air sample preparation:
6.6. XRD Calibration and Analysis
Note: The analytical procedures for XRD and XRF are
written for
Analyze the 24 standards prepared in Section 6.4. according to the XRD SOP (8.15.). Instrument parameters (all angles are in terms of °2Q) for calibration are:
| Ag reference calibration | 38.15° (primary Ag line) 44.33° (secondary Ag line) | |
| ----------------------- V2O5 scan ranges ----------------------- | ||
| Secondary line | 30.40 to 31.70° (quantitative) (31.05° analytical peak) | |
| Primary line | 19.62 to 20.92° (qualitative) | |
| Tertiary line | 25.56 to 26.86° (qualitative) | |
|
| ||
| Scan step size | 0.02° | |
| Integration time | 1 to 10 s | |
| X-ray tube power | 40 kV and 40 mA | |
The primary or secondary Ag line is used as the reference
calibration. The secondary
V2O5 line was
chosen as the quantitative line because interferences from quartz are
present on the primary and tertiary lines. It is possible to make
calibration curves for these two lines only if the
Typical scans of the three recommended analytical lines for a 250 µg V2O5 standard are graphically displayed in Figure 1.
Use available
Consult the XRD SOP (8.15.) for further instructions.
6.7. XRF Calibration and Analysis
- 6.7.1. Direct-beam XRF spectrometer:
6.7.2. Secondary-target XRF spectrometer:
7. Calculations
- 7.1. XRD Data Reduction
The XRD results from line to line may not agree well. Use the 2 line results for calculations unless interferences are present. After calibration and analysis, calculate the microgram quantities of the analyte from the calibration curve. The V2O5 exposure is estimated using the following equation:
Vanadium pentoxide exposure (As V2O5 in mg/m3) = A / B
A = µg
V2O5 found (blank
subtracted)
B = sample air volume in liters
7.2. XRF Data Reduction
Use software features available on instrument:
- 7.2.1. Recall spectrum.
7.2.2. Remove escape peaks.
7.2.3. Remove sum peaks.
7.2.4. Identify V lines and any interfering elements.
7.2.5. Perform background corrections.
7.2.6. Perform profile fit or Gaussian deconvolution of the identified lines.
7.2.7. Tabulate integrated peak counts and masses of standards.
7.2.8. Use the count (or count rate) data to construct a
Compare the XRD and XRF results. As previously mentioned (Section
3.3.),
If a reasonable comparison is obtained between XRD and XRF, report results to the industrial hygienist as mg/m3 V2O5.
Do not report XRD quantitative results; XRF results represent more reliable estimates of the amounts of V2O5 potentially present. Results by XRD represent estimates only.
Disclaimers:
Particulate present on the backup pad constitutes some sample loss. Occasionally this may be seen and can be due to a poor cassette seal on the filter, improper positioning of the filter in the cassette, or poor quality control of the filter and/or cassette. If this type of contamination occurs, relay a note to the compliance officer indicating that some of the sampled material was found on the backup pad and the reported value may be lower than the actual exposure.
8. Reference
- 8.1. Carsey, T.P.: Quantitation of
Vanadium Oxides in Airborne Dusts by
8.2. National Institute for Occupational
Safety and Health: Collaborative Tests of Two Methods for
Determining Free Silica in Airborne Dust (DHHS Publication No.
8.3. National Institute for Occupational
Safety and Health: NIOSH Manual of Analytical Methods, 3rd.
ed., Cincinnati, OH: National Institute for Occupational Safety, 1986.
pp.
8.4. Windholz, M.; ed.: The Merck
Index. 10th ed., Rahway, NJ: Merck and Co., 1983. p.
8.5. American Conference of Governmental
Industrial Hygienists: Documentation of the Threshold Limit
Values and Biological Exposure Indices (Pub. No. ISBN:
8.6. National Institute for Occupational
Safety and Health: Criteria for a Recommended Standard ...
Occupational Exposure to Vanadium (DHEW/NIOSH Pub. No.
8.7. "Air Contaminants; Final Rule" Federal
Register 54:12 (19 Jan. 1989). pp.
8.8. Occupational Safety and Health
Administration Analytical Laboratory: OSHA Manual of Analytical
Methods edited by R.G. Adler (Method No.
8.9. Occupational Safety and Health
Administration Technical Center: Metal and Metalloid
Particulates in Workplace Atmospheres (ICP Analysis) by J.C.
Septon
8.10. Occupational Safety and Health
Administration Technical Center: Vanadium Pentoxide Backup Data
Report
8.11. Occupational Safety and Health
Administration Analytical Laboratory: OSHA Analytical Methods
Manual
8.12. Occupational Safety and Health
Administration Analytical Laboratory: OSHA Analytical Methods
Manual
8.13. American Conference of Governmental
Industrial Hygienists: Threshold Limit Values and Biological
Exposure Indices for
8.14. Occupational Safety and Health
Administration Analytical Laboratory:
8.15. Occupational Safety and Health Administration Analytical Laboratory: Standard Operating Procedure (XRD). Salt Lake City, UT. In progress (unpublished).
8.16. Occupational Safety and Health Administration Analytical Laboratory: Standard Operating Procedure (XRF). Salt Lake City, UT. 1981 (unpublished).
The L lines for the noble gas xenon (Xe) and theshort-lived radioactive element promethium (Pm) were not considered due to their extremely low probability of occurrence in compliance samples.
Analytical Peaks for

Figure
1