| Method no.: | 54 |
| Matrix: | Air |
| Target concentration: | 50 µg/m3 (20 ppb) (OSHA PEL) |
| Procedure: | Samples are collected by drawing a known volume of air through XAD-7 tubes coated with 0.3 mg of 1-(2-pyridyl)piperazine (1-2PP). Samples are desorbed with acetonitrile (ACN) and analyzed by high performance liquid chromatography (HPLC) using a fluorescence or ultraviolet (UV) detector. |
| Recommended air volume and sampling rate: |
15 L at 0.05 L/min |
| Reliable quantitation limit: | 4.8 µg/m3 (1.9 ppb) |
| Standard error of estimate at the target concentration: (Section 4.5.) |
8.0% |
| Special requirements: | The coated XAD-7 tubes should be stored under refrigeration before sampling. |
| Status of method: | Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. |
| Date: April 1985 | Chemists: Donald Burright Duane Lee |
OSHA Analytical Laboratory
Salt Lake City, Utah
1. General Discussion
- 1.1. Background
- 1.1.1. History
In the past there has been no validated sampling and analytical procedures for methyl isocyanate (MIC) in air that could be used for compliance purposes. A search of the literature produced three procedures that were candidates for a validated method:
The first procedure contained only analytical conditions for MIC derivatized with Nitro Reagent (nitrobenzyl-N-n-propylamine). (Ref. 5.1.) No data were presented to indicate the collection efficiency of the sampling device.
The second procedure was a failure report by NIOSH that had derivatized MIC with Nitro Reagent or NMA (naphthylmethylamine) in a toluene impinger. (Ref. 5.2.) Since NIOSH had reported that the reaction time for derivatization was too slow in the impinger and that the derivatization reagents were unstable, NIOSH concluded that the procedure could not be validated.
The third procedure was published by Union Carbide and used a tube packed with a specially treated XAD-2 resin in series with a Cu Cl2 (cupric chloride) bubbler. (Ref. 5.3.) The tube was desorbed with a fluorescamine (Fluram) solution and analyzed by HPLC with a fluorescence detector. Initial attempts to duplicate this procedure were unsuccessful and the procedure was not pursued.
The procedure currently being used for collecting other diisocyanates uses glass fiber filters coated with 1-(2-pyridyl)piperazine (1-2PP). An attempt was made to collect MIC with these filters. The recovery of MIC derivative from the coated filters was low. The second attempted collection procedure used XAD-2 and XAD-7 tubes from SKC which were coated with 1-2PP and tested for collection efficiency of MIC. It was found that higher recoveries were observed with coated XAD-7 than with coated XAD-2. This led to a successful evaluation of the collection of MIC on XAD-7 tubes coated with 1-2PP and analysis on a DuPont Zorbax CN column with a fluorescence detector. A UV detector can be used for this procedure but the detection limit will be higher.
1.1.2. Toxic effects (This section is for information only and should not be taken as a basis for OSHA policy.)
- Inhalation of MIC vapors may cause irritation of the eyes,
nose, throat, and lungs. Cough, shortness of breath, increased
phlegm and chest pains may be present. The liquid splashed in the
eyes may cause permanent damage. The liquid splashed on the skin
may cause irritation. Exposure to MIC may cause a person to become
allergic to it so that extremely low levels of exposure may cause
an asthmatic attack. (Ref. 5.4.)
1.1.3. Operations where exposure may occur
The workers at chemical plants synthesizing MIC may be exposed to it. Also, workers at pesticide manufacturing plant who use MIC are candidates for exposure. MIC is used in the synthesis of several common pesticides including carbaryl, carbofuran, methomyl, and aldicarb. In 1975, over 27 million pounds of MIC were produced and over 25,000 workers could have been exposed to MIC. (Refs. 5.5. and 5.6.)
1.1.4. Physical properties
| CAS no.: | 624-83-9 |
| MW: | 57.05 |
| bp: | 39.1°C at 760 mm Hg |
| mp: | less than -80°C |
| Sp gr: | 0.9599 @ 20°C |
| vp: | 348 mm Hg at 20°C |
| color: | clear, colorless |
| odor: | sharp |
| flash pt.: (open cup) |
less than -18°C |
| synonyms: | isocyanatomethane; isocyanic acid, methyl ester; methylcarbylamine; MIC |
| structure: | H3C-N=C=O |
1.2. Limit defining parameters (The analyte air concentrations listed throughout this method are based on an air volume of 15 L and a solvent desorption volume of 3 mL. Amounts are expressed as the equivalent weight of MIC, even though the MIC derivative was analyzed. Limit defining parameters were determined using a fluorescence detector.)
- 1.2.1. Detection limit of the analytical procedure
The detection limit of the analytical procedure is 0.20 ng per injection of MIC with the fluorescence detector. This is the amount of analyte which will give a peak whose height is about five times the height of the baseline noise. (Section 4.1.)
1.2.2. Detection limit of the overall procedure
The detection limit of the overall procedure is 0.07 µg per sample (4.8 µg/m3 or 1.9 ppb). This is the amount of MIC spiked on the sampling device which allows recovery of an amount of analyte equivalent to the detection limit of the analytical procedure. (Section 4.2.)
1.2.3. Reliable quantitation limit
The reliable quantitation limit is 0.07 µg per sample (4.8 µg/m3 or 1.9 ppb). This is the smallest amount of MIC which can be quantitated within the requirements of a recovery of at least 75% and a precision (±1.96 SD) of ±25% or better. (Section 4.2.)
The reliable quantitation limit and detection limits reported in the method are based upon optimization of the instrument for the smallest possible amount of analyte. When the target concentration of an analyte is exceptionally higher than these limits, they may not be attainable at the routine operating parameters.
- 1.2.4. Sensitivity
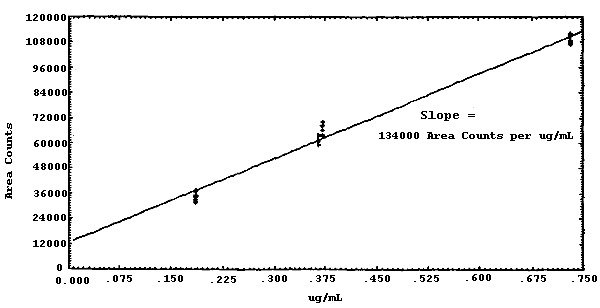
The sensitivity of the analytical procedure over a concentration range representing 0.5 to 2 times the target concentration based on the recommended air volume is 134000 area units per µg/mL. This is determined by the slope of the calibration curve. (Section 4.3.) The sensitivity will vary with the particular instrument used in the analysis.
1.2.5. Recovery
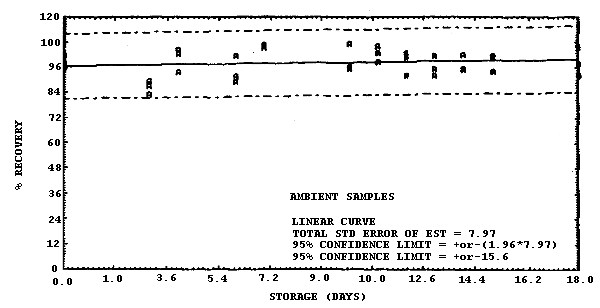
The recovery of MIC derivative from samples used in an 18-day storage test remained above 96.6% when the samples were stored at 22°C. This value was determined from the regression line which was calculated from the ambient data. (Section 4.5.) The recovery of the analyte from the collection medium during storage must be 75% or greater.
1.2.6. Precision (analytical method procedure)
The pooled coefficient of variation obtained from replicate determinations of analytical standards at 0.5, 1, and 2 times the target concentration is 0.040. (Section 4.3.)
1.2.7. Precision (overall procedure)
The precision at the 95% confidence level for the 18-day storage test is ±15.6%. (Section 4.5.) This includes an additional ±5% for sampling error. The overall procedure must provide results at the target concentration that are ±25% or better at the 95% confidence level.
1.2.8. Reproducibility
Six samples, spiked with MIC by liquid injection, and a draft copy of this procedure were given to a chemist unassociated with this evaluation. The samples were analyzed after one day of storage at 22°C. The average recovery was 104.1% with a standard deviation of 3.2%. (Section 4.6.)
1.3. Advantages
- 1.3.1. The analytical procedure is specific and sensitive for
MIC.
1.3.2. The collection system is easier to prepare than the treated XAD-2 tubes of Reference 5.3.
1.4. Disadvantages
- 1.4.1. XAD-7 tubes coated with 1-2PP are not commercially
available.
1.4.2. Due to differences between individual columns, the mobile phase for the HPLC has to have the pH adjusted for every bottle of solvent that is made. The pH affects the retention time of the 1-2PP and the response of the fluorescence detector.
2. Sampling Procedure
- 2.1. Apparatus
- 2.1.1. Samples are collected by use of a personal sampling pump
that can be calibrated to within ±5% of the recommended flow rate
with the sampling device in-line.
2.1.2. XAD-7 tubes from SKC are coated with 0.3 mg of 1-2PP in the following manner. Dissolve the 1-2PP in methylene chloride and place in a separatory funnel. Add 0.05 M sulfuric acid and shake carefully. The 1-2PP is now in the aqueous layer. Separate the layers and discard the organic layer. Make the aqueous layer basic with potassium hydroxide. Extract with methylene chloride and separate the layers. Remove the methylene chloride from the clean 1-2PP using a stream of nitrogen gas. This procedure reduces the contaminant in the 1-2PP that interferes with the HPLC analysis. Make a solution of 1.0 mg/mL of clean 1-2PP in methylene chloride. Open both ends of the XAD-7 tube and with a syringe inject 300 µL of the 1-2PP solution onto the "A" section beads. A vacuum limited by a 0.05 L/min critical orifice is used to draw the solution onto the "B" section. Dry the wet tubes in an unheated vacuum oven for 1 h.
2.1.3. Place plastic caps on the open ends of the tubes and store them at reduced temperature as a precaution to prevent decomposition of the 1-2PP. Exposure to strong sunlight should be avoided.
2.2. Reagents
No sampling reagents are required.
2.3. Sampling technique
- 2.3.1. Attach the coated XAD-7 tube to the sampling pump with
flexible, plastic tubing such that the large, front section of the
sampling tube is exposed directly to the atmosphere. Do not place
any tubing in front of the sampling tube. The sampling tube should
be attached vertically in the worker's breathing zone in such a
manner that it does not impede work performance.
2.3.2. The recommended flow rate is 0.05 L/min with a recommended total air volume of 15 L.
2.3.3. After sampling for the appropriate time, remove the sampling device and install the two plastic caps on the open ends of the tube.
2.3.4. Wrap each sample end-to-end with an OSHA Form 21 seal.
2.3.5. With each set of samples, submit at least one blank. The blank should be handled the same as the other samples except that no air is drawn through it.
2.4. Retention study
A retention study was performed on the MIC derivative by monitoring the effluent from sampling tubes containing only the 80-mg section of coated XAD-7 while drawing air at 0.2 L/min (approximately 80% relative humidity). The XAD-7 tubes had been previously liquid spiked with 1.6 µg of MIC (2 times the target concentration) on the front third of the resin beads. The monitoring was done by analyzing a second tube that had been placed behind the first tube. These backup tubes were changed every hour for 6 h. None of the backup tubes contained any MIC derivative. The front tubes were analyzed and found to contain 95% of the amount spiked on them. Although 0.2 L/min was used in the retention study, this flow rate is too fast for MIC to be derivatized and collected on the XAD-7 tube. The recommended sampling rate is 0.05 L/min, which was determined by studies with gas sampling bags. (Section 4.8.)
2.5. Desorption efficiency
The average desorption efficiency of MIC derivative is 96.1% over the range of 0.5 to 2 times the target concentration. (Section 4.4.)
2.6. Recommended air volume and sampling rate
- 2.6.1. The recommended air volume is 15 L.
2.6.2. The recommended air sampling rate is 0.05 L/min.
2.7. Interferences (sampling)
Any compound that could react with 1-2PP, or compete with it in the reaction to derivatize MIC, should be considered as an interference. Potential interferences include anhydrides, amines, alcohols and carboxylic acids.
2.8. Safety precautions
The sampling equipment should be attached to the worker in such a manner that it will not interfere with work performance or safety.
3. Analytical Procedure
- 3.1. Apparatus
- 3.1.1. High performance liquid chromatograph equipped with an
ultraviolet (UV) or fluorescence detector, manual or automatic
injector, and chart recorder.
3.1.2. HPLC column capable of separating MIC from any interferences. The column employed in this study was a (25-cm × 4.6-mm i.d.) DuPont Zorbax CN (6 µm) column.
3.1.3. An electronic integrator, or some other suitable method of measuring detector response.
3.1.4. Vials, 4-mL with Teflon-lined caps.
3.1.5. Volumetric flasks, pipets, and syringes for preparing standards, making dilutions, and making injections.
3.1.6. Suitable glassware for preparation of MIC urea derivative.
3.1.7. pH meter for adjusting the mobile phase.
3.1.8. Mechanical shaker.
3.2. Reagents
- 3.2.1. Methylene chloride, hexane and acetonitrile, HPLC grade.
3.2.2. Water, HPLC grade. Our laboratory employs a commercially available water filtration system for the preparation of HPLC grade water.
3.2.3. 1-(2-Pyridyl)piperazine, Aldrich.
3.2.4. Methyl isocyanate, K&K.
3.2.5. Ammonium acetate, HPLC grade.
3.2.6. Glacial acetic acid.
3.3. Standard preparation
- 3.3.1. Preparation of purified derivative
A solution containing 0.1 g of MIC in 25 mL of methylene chloride is slowly added to a solution of 0.3 g of 1-2PP in 50 mL of methylene chloride while stirring. The resulting solution is stirred for 1 h. Reduce the volume of methylene chloride to less than 10 mL by evaporation with a stream of dry nitrogen. The solution is added dropwise to 800 mL of hexane while stirring and the resulting precipitate is collected. The precipitate is redissolved in a minimal volume of methylene chloride and reprecipitated in hexane. The precipitate is collected and washed with hexane. The approximate yield is 0.35 g of the derivative after being dried under vacuum. This preparation is a modification of the procedure reported by Goldberg et al. (Ref. 5.7.)
3.3.2. Preparation of standards
A stock standard solution is prepared by dissolving the MIC derivative into ACN. The derivative is expressed as free MIC by multiplying the amount of MIC urea weighed by the conversion factor 0.2590.
Working standards are prepared by diluting the stock standard solutions with ACN.
3.4. Sample preparation
- 3.4.1. The XAD-7 tube is opened and the glass wool plug and the
80-mg "A" section are placed into a 4-mL vial. The 40-mg "B" section
and the two foam plugs are placed into a second 4-mL vial.
3.4.2. Three milliliters of ACN are added to each vial.
3.4.3. A PTFE-lined cap is placed on each vial.
3.4.4. The vials are shaken for 45-60 min.
3.5. Analysis
- 3.5.1. Reverse phase HPLC conditions
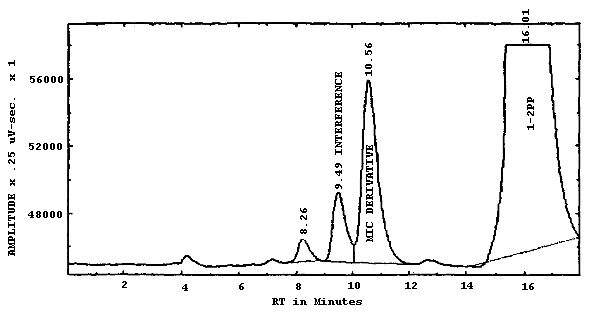
The mobile phase used in this analysis had to be adjusted to optimize the separation on each individual DuPont Zorbax CN column. Although each column was similar at the time of manufacture, the efficiency of each column was affected by the previous analyses that have been performed on the column. Slightly different mobile phases were required by the three Zorbax columns used in this evaluation to obtain the needed separation. The concentration of ACN is varied first to separate the MIC derivative from the interference. Then the pH is adjusted to move the 1-2PP to an acceptable retention time. The increase or decrease of the pH did not substantially affect the separation of the MIC derivative and the interference. The amount of response from the fluorescence detector is decreased as the pH is lowered.
| column: | 25-cm × 4.6-mm i.d. stainless steel column packed with 6 µm DuPont Zorbax CN |
| mobile phase: | 0.005-0.02 M ammonium acetate in 20-25%
|
| flow rate: | 0.8-1.0 mL/min |
| fluorescence detector: | 240 nm excitation 370 nm emission |
| UV detector: | 254 nm |
| injection size: | 5-20 µL |
| retention time: | 8-12 min |
| chromatogram: | Figure 3.5.1. |
3.5.2. Alternate conditions
| column: | 12.5-cm × 4-mm i.d. Hibar LiChroCART packed with 7-µm LiChrosorb RP-8 |
| mobile phase: | 0.01 M ammonium acetate in 12% ACN/88% water (v/v) |
| flow rate: | 1.0 mL/min |
| fluorescence detector: | Same as above |
| UV detector: | Same as above |
| injection size: | Same as above |
| retention time: | 12 min |
3.5.3. An external standard procedure is used to prepare a calibration curve using at least two stock solutions from which working standards are made. The calibration curve is prepared daily. The samples are bracketed with analytical standards.
3.6. Interferences (analytical)
- 3.6.1. Any compound having the same retention time as the MIC
derivative is an interference. Generally, chromatographic conditions
can be altered to separate an interference from the analyte.
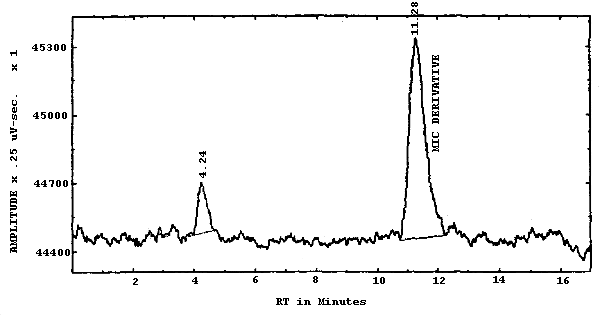
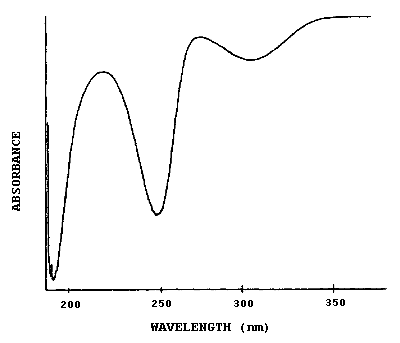
3.6.2. Retention time on a single column is not proof of chemical identity. Analysis by an alternate HPLC column, absorbance response ratioing, and mass spectrometry are additional means of identification. (See UV spectrum for MIC derivative. Figure 4.7.)
3.7. Calculations
The concentration in µg/mL of MIC present in a sample is determined from the detector response of the analyte. Comparison of sample response with a least squares curve fit for standards allows the analyst to determine the concentration of MIC in µg/mL for the sample. Since the sample volume is 3 mL, the results in µg/m3 of air are expressed by the following equation:
3.8. Safety precautions
- 3.8.1. Avoid exposure to the MIC standards.
3.8.2. Avoid skin contact with all solvents.
3.8.3. Wear safety glasses at all times.
4. Backup Data
- 4.1. Detection limit of the analytical procedure
The detection limit of the analytical procedure is 0.20 ng per injection and was determined by injecting 10 µL of a 0.02 µg/mL standard. This amount produced a peak whose height was about 5 times the height of the baseline noise. The injection volume recommended in the analytical procedure (10 µL) was used in the determination of the detection limit for the analytical procedure. (Figure 4.1.)
4.2. Detection limit of the overall procedure and reliable quantitation limit
The detection limit of the overall procedure and the reliable quantitation limit are 0.072 µg MIC per sample (4.8 µg/m3 or 1.9 ppb based on a 15-L air volume). Six samples were prepared by injecting 15 µL of a MIC solution (4.79 µg/mL) onto coated XAD-7 tubes. The samples were stored overnight at room temperature. The samples were analyzed the following day and the percent recovery is reported in Table 4.2.
Data for Detection Limit of the Overall
Procedure and Reliable Quantitation Limit
|
| ||||||
| sample no. |
µg spiked |
µg found |
% recovery | |||
|
| ||||||
| 1 2 3 4 5 6 |
0.0718 0.0718 0.0718 0.0718 0.0718 0.0718 |
0.0542 0.0578 0.0590 0.0630 0.0603 0.0597 |
75.5 80.4 82.1 87.7 84.0 83.1 | |||
| ||||||
|
| ||||||
4.3 Sensitivity and precision (analytical method only)
The following data were obtained from multiple injections of analytical standards. The data are also presented graphically in Figure 4.3.
MIC Derivative Sensitivity and Precision Data
|
| ||||
| × target conc. µg/mL |
0.5× 0.186 |
1× 0.366 |
1× 0.372 |
2× 0.732 |
|
| ||||
| area counts SD CV |
31678.2 37681.2 36815.4 35414.2 35037.9 33358.5 34463.7 32735.5 34648.1 2028.8 0.0586 |
59532.5 61035.5 61602.6 59641.0 59691.1 63773.4 62320.5 64705.6 61537.8 1961.5 0.0319 |
66397.0 63941.7 68941.5 63990.2 68327.0 69005.1 63979.2 70268.4 66856.3 2617.8 0.0392 |
111699 107611 106980 108987 111028 112342 107361 108279 109285.9 2108.8 0.0193 |
|
| ||||
The pooled coefficient of variation for MIC is 0.0399. The sensitivity of MIC is 134000 area counts per µg/mL.
4.4. Desorption efficiency
The following data represent the analysis of eighteen coated XAD-7 tubes that were liquid spiked with MIC. The average desorption efficiency for coated XAD-7 tubes is 96.1%.
Desorption Efficiency of MIC Derivative
|
| |||
| × target conc. µg/sample |
0.5× 0.399 |
1× 0.798 |
2× 1.596 |
|
| |||
| desorption efficiency, % SD |
91.7 92.0 91.8 91.9 95.7 95.4 93.08 1.92 |
92.2 95.2 95.4 96.0 94.7 109.2 97.12 6.06 |
96.3 97.2 95.7 98.5 96.5 104.5 98.12 3.27 |
|
| |||
4.5. Storage data
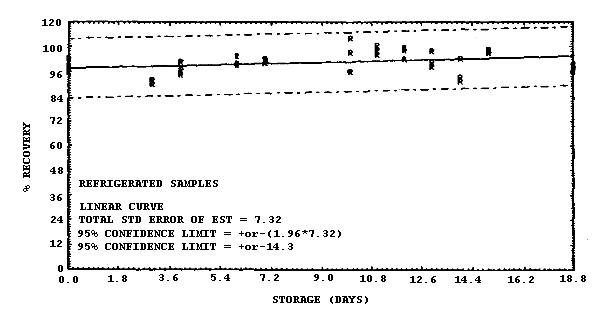
Sixty-nine samples were generated by liquid spiking 0.798 µg of MIC on the glass wool plug inside each coated XAD-7 tube. The tubes then had 3 L of air (at approximately 80% relative humidity) pulled through them. Three samples were analyzed immediately after generation, 33 were stored in a refrigerator at 4°C and 33 were stored in a closed drawer at 21°C. The results of recovery versus storage time are given in Table 4.5. and shown graphically in Figures 4.5.1. and 4.5.2.
Storage Tests
|
| |||||||
| storage time | % recovery | ||||||
| (days) | (refrigerated) | (ambient) | |||||
|
| |||||||
| 0 3 4 6 7 10 11 12 13 14 15 18 |
98.9 89.7 94.4 103.8 102.0 105.6 104.6 107.6 105.9 102.6 106.8 96.3 |
95.9 89.7 100.9 99.1 100.0 96.0 106.4 102.2 98.6 93.4 106.2 99.5 |
102.4 91.6 97.0 100.2 101.2 112.3 108.6 106.2 100.6 91.4 105.1 97.8 |
102.4 83.1 93.7 101.7 105.7 96.7 99.0 100.7 101.7 102.2 101.0 106.0 |
95.9 89.6 102.6 89.2 106.9 95.8 103.4 102.8 95.5 94.9 101.5 98.0 |
98.9 87.1 104.4 91.6 106.8 107.6 106.3 92.0 92.1 95.2 94.0 92.7 | |
|
| |||||||
4.6. Reproducibility data
Six samples, liquid spiked with 0.768 µg of MIC, were given to a chemist unassociated with this study. The samples were analyzed after being stored for 1 day at ambient temperature. The results are presented below and are corrected for desorption efficiency.
Reproducibility Results
|
| |||
| % recovery | statistics | ||
|
| |||
| 108.1 106.1 102.0 100.0 106.1 102.0 |
SD |
= = |
104.1 3.2 |
|
| |||
4.7. UV spectrum
Figure 4.7. is the UV spectrum in ACN of the 1-2PP derivative of MIC used in this study.
4.8. Gas sampling bag study
A gas sampling bag was filled with 150 L of dry air. Eight microliters of a solution of MIC (0.94 mg/mL in methylene chloride) was injected into the bag, resulting in a theoretical concentration of 50 µg/m3. This test atmosphere was then sampled side-by-side at 0.05 L/min with four XAD-7 tubes coated with 1-2PP and at 0.1 L/min with two XAD-7 tubes coated with Nitro Reagent. The average concentration found on the tubes coated with 1-2PP was 56 µg/m3, which was greater than the average concentration obtained from the Nitro Reagent tubes, 51 µg/m3. The Nitro Reagent coated XAD-7 tubes were used to cross check the accuracy of the gas sampling bag dilution procedure. Although it had been reported that Nitro Reagent was too unstable and did not react fast enough to derivatize MIC in impingers, our preliminary studies showed that Nitro Reagent coated on XAD-7 would collect MIC and that the derivative could be chromatographed successfully. If the derivatizing reagent 1-2PP had not worked, the Nitro Reagent procedure probably could have been evaluated.
Sampling at 0.2 L/min was tested with a 50 µg/m3 gas sampling bag and by liquid spiking tubes with 0.798 µg of MIC. During both tests, the 1-2PP coated tubes did not collect or retain the airborne MIC as efficiently as they did at 0.05 L/min. The collection efficiency was about 66% at the higher flow rate which is unacceptable. Therefore the recommended sampling rate is 0.05 L/min.
5. References
- 5.1. Lessley, S.D.; Nelson, J.H. Utah Biological Testing
Laboratory, 1979, unpublished results.
5.2. Methyl Isocyanate (MIC) Failure Report No. S252, prepared under NIOSH Contract No. CDC-99-74-45, 1979.
5.3. "Analytical Techniques in Occupational Health Chemistry", Dollberg, D.D.; Verstuyft, A.W., Eds.; American Chemical Society, Washington D.C., 1980.
5.4. "Occupational Health Guidelines for Chemical Hazards" NIOSH/OSHA, Jan. 1981, DHHS(NIOSH) Publication No. 81-123.
5.5. Toxicology Data Bank (Online Computerized Database), National Library of Medicine; DHHS, Rockville, MD.
5.6. "Information Profiles on Potential Occupational Hazards", Hoecker, J.E.; Durkin, P.R.; Hanchett, A.; Davis, L.N.; Meylan, W.M.; Bosch, S.J. Syracuse Research Corporation: New York, 1977.
5.7. Goldberg, P.A.; Walker, R.F.; Ellwood, P.A.; Hardy, H.L. J. Chromatogr. 1981, 212, 93.