(This method was fully evaluated with
Stoddard solvent. It can also be used to
determine V.M.&P. naphtha and mineral spirits.)
| Method no.: | 48 |
| Matrix: | Air |
| Target concentration: | 2900 mg/m3 Stoddard solvent (OSHA PEL) |
| Procedure: | Samples are collected by drawing a known volume of air through charcoal tubes. Samples are desorbed with carbon disulfide (CS2) and analyzed by gas chromatography (GC) using a flame ionization detector (FID). |
| Recommended air volume and sampling rate: | 3 L at 0.2 L/min |
| Reliable quantitation limit: | 0.77 mg/sample (260 mg/m3) |
| Precision: (1.96 SD) (Section 4.3.2.) |
17.8% |
| Status of method: | Evaluated method. This method has been subjected to the established evaluation procedures of the Organic Methods Evaluation Branch. |
| Date: November 1984 | Chemist: Michael L. Shulsky |
OSHA Analytical Laboratory
Salt Lake City, Utah
1. General Discussion
- 1.1. Background
- 1.1.1. History
Three refined petroleum mixtures are routinely analyzed at this laboratory. They are Stoddard solvent (boiling range 160-210°C), mineral spirits (boiling range 150-200°C), and petroleum distillates (V.M.&P. naphtha; boiling range 95-160°C). These mixtures will collectively be termed petroleum distillate fractions (PDF) throughout this method. All of these PDFs contain aliphatic and to a lesser extent aromatic hydrocarbons. (Ref. 5.1.)
The procedures for collection (charcoal tubes) and analysis
(GC/FID) of PDFs described in this evaluation are basically those
used in NIOSH methods S380 and S382. (Ref. 5.2.) For preparation of
analytical standards, these NIOSH methods require a sample of the
bulk material presumed to be the source of the air contamination
(this bulk material will be referred to as the "source PDF"
throughout this method). The shipment of source PDFs, which are
often flammable, is inconvenient and the materials sometime require
distillation before use in standards. For these reasons and because
similar responses to different hydrocarbons are observed using a FID
(Ref. 5.3.), the use of analytical standards prepared from a PDF
which is not the source PDF was investigated. In order to determine
analytical conditions, it was assumed that this substitute PDF
("
Internal standards (Istd) are routinely used in solvent analyses at this laboratory. Since the actual constituents of PDFs are unknown, the presence of an internal standard may cause an interference with the PDF or unduly lengthen the analysis time. For these reasons, the possibility of using an external standard (Estd) procedure was examined.
Also, in preliminary work it became apparent that the manner in which the baseline was set was a concern. If the data system was allowed to automatically set the baseline, inconsistencies in the positions to which the baseline was drawn were noticed (Figures 4.8.1. and 4.8.2.). This produced calibration errors at lower concentrations of PDFs. To overcome this problem, an evaluation of certain "integrate functions" available in the data system software which control the baseline was done (Section 4.8.4.).
In order to evaluate the parameters of baseline, Estd, and
material used to prepare analytical standards, a study was done
utilizing eight different PDFs consisting of five Stoddard solvents,
two V.M.&P. naphthas and one mineral spirits. These were used to
spike 8 sets of 12 charcoal tubes. Each 12-tube set was quantitated
using analytical standards prepared from both source and
The results of this study indicate several things; there is no
significant difference in results obtained by using either the
source or
Other tests performed for this evaluation were break through, storage stability, desorption efficiencies, precision of the analytical procedure, sensitivity and reliable quantitation limit. The breakthrough tests were performed with both a Stoddard solvent (Section 4.4.1.) and a V.M.&P. naphtha (Section 4.4.2.) to ensure the collection procedure would work for the more volatile constituents of a V.M.&P. naphtha. All of the other tests were performed using a Stoddard solvent but the collection and analytical procedure should also be applicable to petroleum distillates and mineral spirits.
There are two OSHA PELs that pertain to petroleum distil late fractions. The PELs are 2900 mg/m3 for Stoddard solvent and 2000 mg/m3 for petroleum distillates (naphtha). Due to numerous synonyms and the overlapping boiling range fractions that are available, there is much confusion as to which standard is applicable in many instances. Mineral spirits, which is almost identical to Stoddard solvent in boiling range, should be compared to the Stoddard solvent PEL; while the lower boiling range petroleum distillate fractions should be compared to the petroleum distillate (naphtha) PEL.
This evaluation shows that PDFs can be collected using charcoal
with a 3-L air volume, analyzed by GC/FID and a
1.1.2. Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy).
"Short-term Exposure: Overexposure to Stoddard solvent causes irritation of the eyes, nose, and throat and may cause dizziness. Very high air concentrations may cause unconsciousness and death. Long-term Exposure: Prolonged overexposure to the liquid may cause skin irritation." (Ref. 5.4.)
"Short-term Exposure: Overexposure to petroleum distillates may cause dizziness, drowsiness, headache, and nausea. They may also cause irritation of the eyes, throat, and skin. Long-term Exposure: Prolonged exposure may cause drying and cracking of the skin." (Ref. 5.5.)
Men were exposed to mineral spirits concentrations of 2500 to 5000 mg/m3 for an unspecified time period. Both concentrations produced nausea and vertigo in the subjects. In another study at 4000 mg/m3 there was a prolongation of reaction time. (Ref. 5.1.)
1.1.3. Potential workplace exposure
- NIOSH estimates that about 600,000 workers in the United
States are potentially exposed to all "specialized naphthas" (Ref.
5.1.).
Petroleum distillates (V.M.&P. naphtha) is used as a quick evaporating paint thinner. Stoddard solvent is used in the dry cleaning industry. Mineral spirits is a general purpose thinner, a dry cleaning agent, and a solvent for paint and varnish industries. (Ref. 5.1.)
1.1.4. Physical properties (Ref. 5.1. unless otherwise stated)
| Petroleum distillates | |
| molecular weight: | approximately 87-114 |
| odor: | pleasant aromatic odor |
| boiling range: | 95 - 160°C |
| specific gravity: | 0.7275 - 0.7603 |
| color: | clear, water white to yellow |
| vapor pressure: | 2 - 20 mm Hg at 20°C |
| flashpoint: | -6.7 to 12.8°C (closed cup) |
| synonyms: | benzine, naphtha 76, ligroin, high boiling petroleum ether |
| molecular species: | C7-C11 |
| Stoddard solvent | |
| molecular weight: | approximately 135 - 145 |
| odor: | kerosene-like |
| boiling range: | 160 - 210°C |
| specific gravity: | 0.75 - 0.80 |
| color: | colorless |
| vapor pressure: | 4 - 4.5 mm Hg at 25°C |
| flashpoint: | 37.8°C (closed cup) |
| synonyms: | 140 flash solvent, odorless solvent and low end point solvent |
| molecular species: | C9-C11 |
| Mineral spirits | |
| molecular weight: | approximately 144 - 169 |
| odor: | pleasant sweet odor |
| boiling range: | 150 - 200°C |
| specific gravity: | 0.77 - 0.81 |
| color: | clear, water white |
| vapor pressure: | 0.8 mm (Hg) at 20°C |
| flashpoint: | 30.2 - 40.5°C (closed cup) |
| synonyms: | white spirits, petroleum spirits, and light petrol |
| molecular species: | C9-C12 |
1.2. Limit defining parameters (Air concentrations are based on the recommended air volume (3 L) and a desorption volume of 1 mL.)
- 1.2.1. Detection limits
Since PDF consist of numerous and varying components, the determination of meaningful detection limits was not considered feasible.
1.2.2. Reliable quantitation limit
The reliable quantitation limit is 0.77 mg/sample (260 mg/m3) This concentration was arrived at by taking all the results for calibration methods #4 and #5 from Tables 4.8.1. through 4.8.8. that were near certain concentrations, i.e. 0.3 mg/mL and 0.7 mg/mL, and finding the average recoveries, the average concentrations, and standard deviations (SD) near those concentrations. The results for samples near 0.77 mg/mL met both the requirements of 75% recovery and a precision (1.96 SD) of ±25% or better. (Section 4.2.)
1.2.3. Sensitivity
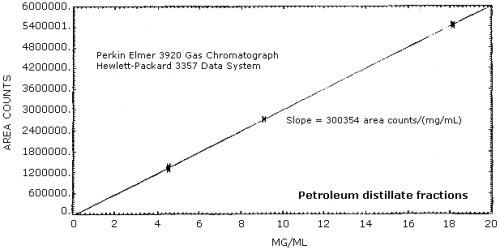
The sensitivity of the analytical procedure over a range representing 0.5 to 2 times the target concentration based on the recommended air volume is 300954 area units per mg/mL. This is determined by the slope of the calibration curve. (Section 4.3.3.)
1.2.4. Recovery
The recovery of samples used in a 15-day storage test remained above 94% (Section 4.6.). The recovery of the analyte from the collection medium during storage must be 75% or greater.
1.2.5. Precision of the analytical procedure
The pooled coefficient of variation obtained from replicate determinations of analytical standards at 0.5, 1, and 2 times the target concentration is 0.019 (Section 4.3.1.).
1.2.6. Precision of the overall procedure
The precision of the overall procedure at the 95% confidence level is ±17.8% (Section 4.3.2.). This includes an additional 5% for sampling error. The overall procedure must provide results that are ±25% or better at the 95% confidence level.
1.2.7. Reproducibility
Six samples spiked by liquid injection and a draft copy of this procedure were given to a chemist unassociated with this evaluation. The samples were analyzed after 2 days of storage at 22°C. The average recovery was 97.7% with a SD of ±3.53%. (Section 4.7.)
1.3. Advantages
- 1.3.1. The collection procedure is convenient.
1.3.2. The analytical procedure is rapid and precise.
1.4. Disadvantages
None
2. Sampling Procedure
- 2.1. Apparatus
- 2.1.1. A personal sampling pump which can be calibrated within
±5% of the recommended flow rate is needed.
2.1.2. Coconut shell charcoal tubes which consist of glass tubes 7 cm long, 6-mm o.d., and 4-mm i.d., containing a 100-mg section and a 50-mg section of charcoal separated with a urethane foam plug are used. The glass tube is flame sealed at both ends. For this evaluation, SKC, Inc. charcoal tubes, lot 120, were used.
2.2. Reagents
None required
2.3. Technique
- 2.3.1. Immediately before sampling, break open the ends of the
charcoal tube. All tubes should be from the same lot of charcoal.
2.3.2. Connect the charcoal tube to the pump with a short piece of flexible tubing. The 50-mg portion of the charcoal tube is used as the backup section; therefore, air should flow through the 100-mg portion first.
2.3.3. Position the tube vertically to avoid channeling through the charcoal.
2.3.4. Air being sampled should not pass through any hose or tubing before entering the charcoal tube.
2.3.5. Record the temperature and relative humidity of the atmosphere being sampled.
2.3.6. Immediately after sampling, seal the ends of the tubes with the plastic caps.
2.3.7. With each set of samples, submit at least one blank charcoal tube from the same lot as the sample tubes. The blank tube should be treated in the same manner as the samples (break ends, seal, transport) except no air is drawn through it.
2.3.8. Transport the samples and corresponding paperwork to the laboratory for analysis.
2.3.9. Submit source PDF whenever possible. Place the material in glass bottles with Teflon-lined caps, and transport to laboratory separately from air samples.
2.4. Breakthrough
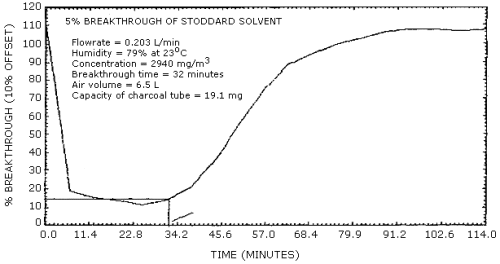
Studies to determine the 5% breakthrough value were done near the PEL for Stoddard solvent, using a dynamically generated atmosphere with approximately 75% relative humidity at 22°C and a sampling rate of 0.203 L/min. These studies were performed using only the 100 mg portion of a charcoal tube. The average breakthrough for Stoddard solvent was 6.9 L and average capacity was 20 mg. (Section 4.4.1.). Breakthrough studies were performed with a petroleum distillate (V.M.&P.) naphtha since this type of PDF boils at a lower temperature. The average breakthrough volume for this V.M.&P. naphtha was 9.4 L and the average capacity was 20.3 mg. (Section 4.4.2.)
2.5. Desorption efficiency
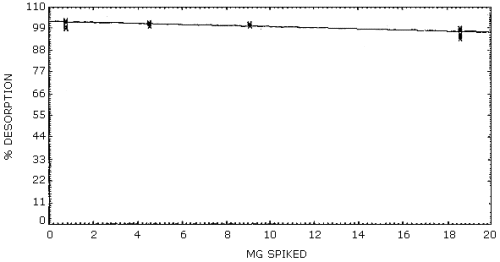
Desorption efficiencies were determined at several different loadings of Stoddard solvent. These loadings corresponded to the mass of Stoddard solvent which would be collected on a charcoal tube when sampling 3 L of an atmosphere containing 0.1, 0.5, 1, and 2 times the PEL. The tubes were prepared by liquid injection of the Stoddard solvent and stored in a refrigerator for 24 h before analysis. The average desorption efficiency was 100%. (Section 4.5.)
2.6. Recommended air volume and sampling rate.
The recommended air volume is 3 L at 0.2 L/min.
2.7. Interferences
- 2.7.1. Since charcoal will collect vapors from many organic
compounds all organics being used in significant amounts near the
sampling area could decrease the capacity of the charcoal for PDF.
2.7.2. Water vapor also may decrease the capacity of charcoal.
2.8. Safety precautions
- 2.8.1. Wear eye protection when breaking the ends of the
charcoal tubes.
2.8.2. Place the sampling pump on the employee in a manner so it will not interfere with the work being done.
2.8.3. Place the charcoal tube in a holder so the broken ends are not exposed.
2.8.4. Obey all safety regulations of the workplace.
3. Analytical Procedure
- 3.1. Apparatus
- 3.1.1. A gas chromatograph (GC) equipped with a flame ionization
detector (FID) is used for analysis. A Hewlett-Packard 5710 GC was
primarily used in this evaluation.
3.1.2. A GC column capable of separating carbon disulfide (CS2) and the internal standard, if any, from the constituents of the PDF. For this evaluation, a 20 ft by 1/8 in. stainless steel column packed with 10% SP-1000 on 80/100 Supelcoport was used.
3.1.3. An integrator for determining peak area is needed. A Hewlett-Packard 3357 data system was used.
3.1.4. Small vials with Teflon-lined caps for desorption of charcoal: Two-milliliter vials are preferable.
3.1.5. Microliter syringes such as 10-µL for preparing standards and 1-µL for sample injection are needed.
3.1.6. Pipettes for dispensing the desorbing solution may be used. A 1-mL reagent dispenser is convenient.
3.1.7. Volumetric flasks are used for standard preparation.
3.1.8. An analytical balance is used to prepare standards.
3.1.9. A distillation apparatus may be needed.
3.2. Reagents
- 3.2.1. Carbon disulfide, reagent grade.
3.2.2. Source PDF, when possible, from the operation where sampling was done.
3.2.3. Internal standard compound such as hexylbenzene, reagent grade (optional).
3.2.4. GC grade hydrogen, air and nitrogen.
3.2.5. Desorbing solvent: CS2 or 1 µL internal standard/mL CS2.
3.3. Standard preparation
- 3.3.1. Analytical standards are prepared in the desorbing
solvent.
3.3.2. Source PDF received from the sampling site may be used as the analytical standard if it appears clear and colorless, and has a density in the range of 0.74-0.79 g/mL. If the bulk is colored or has a density greater than 0.79 g/mL, it needs to be distilled to separate the volatile solvents from the pigments or heavier oils before it can be used as an analytical standard.
3.3.3. If source PDF is not submitted or is unusable, a nonsource PDF from the laboratory should be used.
3.3.4. Standards must be prepared at four different concentrations so proper integration of the peaks may be confirmed (Section 3.5.3.). A useful range for standard concentrations is approximately 1 µL/mL to 10 µL/mL.
3.4. Sample preparation
- 3.4.1. The 100-mg portion of the charcoal tube is placed in a
vial and the 50-mg portion is placed in a separate vial. The glass
wool and urethane plugs are discarded.
3.4.2. One milliliter of desorbing solvent is added to each vial.
3.4.3. The vials are immediately capped and shaken periodically for 30 min before analysis.
3.5. Analysis
- 3.5.1. GC conditions
| oven: | initial temperature 100°C for 4 min programmed to 180°C at 8°/min |
| injector: | 200°C |
| detector: | 225°C |
| nitrogen (carrier): | 22 mL/min |
| hydrogen: | 30 mL/min |
| air: | 250 mL/min |
| injection size: | 1 µL |
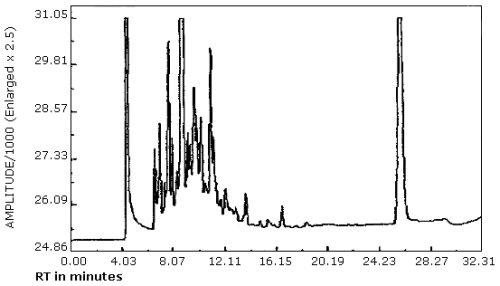
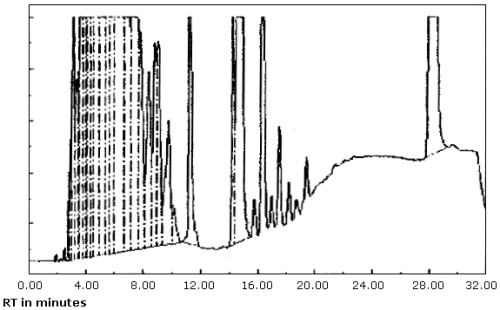
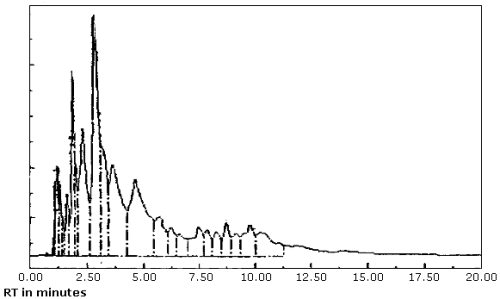
| chromatogram: | Figure 3.5.1. |
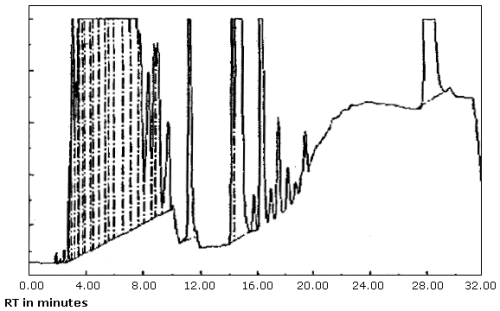
3.5.2. The data system used in this evaluation was a Hewlett-Packard 3357 which contains several "integrate functions." The integrate function termed "hold the baseline" should be used for the analyses. This function should be started before the constituents of the petroleum distillate fraction begin to elute from the column and it should be canceled after the PDF constituents have eluted or when column bleed becomes significant whichever occurs first.
3.5.3. The areas of the peaks due to PDF constituents are added together (area summation) in the analysis of the standards and samples. The summed areas and the concentration of the analytical standards are used to determine a linear least squares fit equation. The concentration of the samples is determined by entering their summed areas into the least squares equation.
3.5.4. If the peaks present in the samples do not elute in approximately the same time range as the standards, a comparison of the constituents in the samples and standard should be done by GC/MS to confirm that the samples do contain PDF type compounds and of what type for reporting purposes. If distinct analytes are confirmed by GC/MS, their identity and approximate concentration should be reported.
3.5.5. Any sample above the PEL should be confirmed by GC/MS or another suitable technique.
3.6. Interferences
- 3.6.1. Since PDF are mixtures of aliphatic and aromatic
hydrocarbons and elute from a GC in a peak cluster, it may be
difficult to eliminate interfering compounds. If a large interfering
peak appears in an air sample, identification by GC/MS may be
necessary.
3.6.2. It may be difficult to separate a single analyte which is requested for analysis from the PDF constituents. Changing columns such as from a polar to a non-polar (SP-1000 to an SP-2100) may help separate the analyte.
3.7. Calculations
- 3.7.1. PDF should be reported as mg/m3
since any ppm value would require the use of an approximate
molecular weight.
3.7.2. The air concentration in mg/m3 is determined from the mass of analyte in the sample as in the following example:
- Upon analysis, 3.5 mg was found for a sample with a 3-L air
volume.
mg/m3 = (mg/desorption
efficiency)/air vol.
mg/m3 = (3.5
mg/1.00)/(0.003
m3)
mg/m3 =
1167 mg/m3
3.8. Safety precautions
- 3.8.1. Work in a hood when using solvents during sample and
standard preparation.
3.8.2. Keep solvents away from sources of high temperatures such as detectors and injectors.
3.8.3. Avoid skin contact with solvents.
3.8.4. Wear safety glasses at all times.
4. Backup data
- 4.1. Detection limits of the analytical and overall procedure
The determination of detection limit values is not practical in the context of a rigid definition such as a peak with a height of 5 times the baseline noise. Since PDFs may have similar constituents which have unsimilar concentrations, there is no one representative peak that can be used to determine detection limits for all PDFs.
4.2. Reliable quantitation limit
The amount of 0.77 mg/sample (260 mg/m3) is determined to be the approximate amount reliably quantitated for any applicable petroleum distillate fraction within the requirements of at least 75% recovery and a precision (1.96 SD) of ±25% or better. The injection size recommended in the analytical procedure (1 µL) was used in the determination of the reliable quantitation limit.
Reliable Quantitation Limit Data
|
| ||||||||||||||
| sample | calibration | mass (mg) | mass (mg) | % | ||||||||||
| number | method* | Istd | spiked | recovered | recovered | |||||||||
|
| ||||||||||||||
| 1 8 14 21 31 35 39 47 51 60 65 70 76 83 |
#4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 #4 #5 |
yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no yes no |
0.789 0.789 0.789 0.789 0.789 0.789 0.789 0.789 0.777 0.777 0.777 0.777 0.777 0.777 0.777 0.777 0.753 0.753 0.753 0.753 0.753 0.753 0.753 0.753 0.754 0.754 0.754 0.754 0.754 0.754 0.754 0.754 0.779 0.779 0.779 0.779 0.779 0.779 0.779 0.779 0.761 0.761 0.761 0.761 0.761 0.761 0.761 0.761 0.776 0.776 0.776 0.776 0.776 0.776 0.776 0.776 |
0.873 0.823 0.773 0.762 0.847 0.806 0.751 0.746 0.812 0.779 0.930 0.863 0.753 0.778 0.845 0.845 0.643 0.663 0.703 0.689 0.684 0.696 0.748 0.723 0.658 0.552 0.602 0.529 0.655 0.715 0.609 0.685 0.828 0.823 0.825 0.821 0.820 0.810 0.818 0.809 0.793 0.778 0.816 0.788 0.824 0.793 0.831 0.819 0.900 0.949 0.838 0.845 0.851 0.912 0.792 0.815 |
111 104 98 96 107 102 95 95 104 100 120 111 97 100 109 109 85 88 93 92 91 92 99 96 87 73 80 70 87 95 81 91 106 106 106 105 105 104 105 104 104 102 107 102 108 104 109 108 116 122 108 109 110 117 102 105 | |||||||||
| ||||||||||||||
|
| ||||||||||||||
| * Explanation of calibration methods under Table 4.8.2. | ||||||||||||||
4.3. Precision and Sensitivity
- 4.3.1. The precision of the analytical method was determined by
replicate injections of analytical standards prepared at 0.5, 1, and
2 times the target concentration. The pooled coefficient of
variation is 0.019.
Precision of Analytical Method
|
| |||
| × target conc. | 0.5× | 1× | 2× |
|
| |||
| area counts | 1322304 | 2761497 | 5482172 |
| 1272435 | 2731651 | 5394150 | |
| 1328744 | 2757576 | 5505614 | |
| 1350244 | 2735224 | 5451850 | |
| 1377105 | 2731653 | 5466193 | |
| 1381708 | 2693328 | 5413149 | |
| 1338756 | 2735155 | 5452188 | |
| SD | 40538 | 24375 | 42052 |
| CV | 0.030 | 0.0089 | 0.0077 |
|
| |||
4.3.2. The precision of the overall procedure was calculated by taking the average of the SDs for methods #4 and #5 (both Istd and Estd) from Table 4.8.1. and multiplying by 1.96. This number includes ±5% for sampling error. The usual value on the cover page is the standard error of estimate from the storage test but in this evaluation this value would not have included variability for using different PDFs for analytical standards.
4.3.3. Sensitivity is defined as the slope of the calibration curve for analytical standards from 0.5 to 2 times the target concentration. (Table 4.3.1., Figure 4.3.2.) The sensitivity is 300954 area counts/(mg/mL). The sensitivity will change depending on the detector and method of integration.
4.4. Breakthrough
- 4.4.1. Breakthrough was determined by sampling a dynamically
generated test atmosphere of Stoddard solvent (about 2900
mg/m3 with 76% RH at 23°C), using a
charcoal tube containing only the 100-mg portion of charcoal and
monitoring the concentration of Stoddard solvent in the air which
had passed through the charcoal.
4.4.2. Breakthrough tests were also performed using a petroleum distillate bulk since its boiling range is lower than Stoddard solvent and it contains more volatile constituents. The test atmospheres were about 2000 mg/m3 with 74% RH at 23°C. Three tests were performed, with 5% breakthrough air volumes of 9.6, 9.1 and 9.5 L and capacities of 20.82, 19.73 and 19.95 mg being obtained respectively. The average capacity was 20.3 mg and the average 5% breakthrough air volume was 9.4 L.
4.5. Desorption efficiencies
Desorption efficiencies were determined by injecting known amounts of Stoddard solvent onto the 100-mg portion of six charcoal tubes, allowing them to sit overnight and analyzing the tubes on the next day. The average desorption efficiency over the range of 0.08 to 2 times the target concentration is 100%.
Desorption Efficiencies
|
| ||||
| × target conc. µg/sample |
0.08× 0.76 |
0.5× 4.55 |
1× 9.1 |
2× 18.6 |
|
| ||||
| desorption efficiency, % |
103 102 99 102 100 103 102 |
100 101 102 102 101 101 101 |
100 100 100 101 101 101 101 |
99 99 98 95 96 94 97 |
|
| ||||
4.6. Storage data
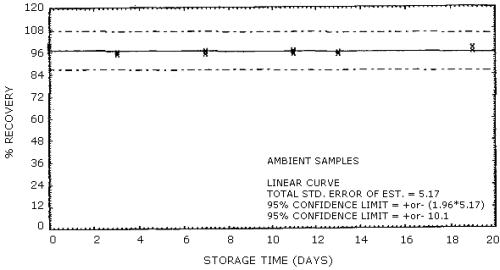
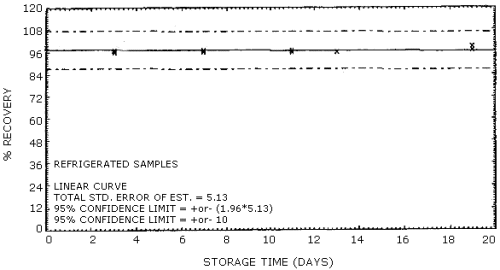
Thirty-six samples were collected from a dynamically generated atmosphere of Stoddard solvent. The atmosphere was approximately 2900 mg/m3 and 75% RH at 22°C. Of these 36 samples, six were analyzed immediately, while the remaining 30 were stored; 15 at ambient temperature and 15 at -5°C. Approximately every third day, 3 samples from each of the storage sets were analyzed. The average recovery was 96% for ambient storage and 97% for refrigerated storage. The data of Table 4.6. are shown graphically in Figures 4.6.1. and 4.6.2.
Storage Tests
|
| ||||||
| storage time (days) |
%
recovery (refrigerated) |
%
recovery (ambient) | ||||
|
| ||||||
| 0 3 7 11 13 19 |
99 96 96 97 96 97 |
99 97 97 96 96 99 |
99 96 97 96 96 97 |
97 95 95 95 95 98 |
99 96 96 96 96 96 |
100 96 97 97 96 96 |
|
| ||||||
4.7. Reproducibility data
Six samples, spiked by liquid injection, and a draft copy of this procedure were given to a chemist unassociated with this evaluation. The samples were analyzed after 3 days of storage at 22°C. The average recovery was 97.7% with a standard deviation of ±3.53%.
Reproducibility Results
|
| |||||
| amount spiked (µg) | amount recovered (µg) | % recovered | |||
|
| |||||
| 7756 7756 7756 7756 7756 7756 |
7432 7510 7443 7493 7466 8136 |
95.8 96.8 95.8 96.6 96.3 104.9 | |||
| |||||
|
| |||||
4.8. Quantitation factors
- 4.8.1. A total of 96 samples were used to evaluate differences
between source and
4.8.2. The six analytical standards were analyzed at the same
time as the samples. A linear least squares fit for each set of
standards was used in all of the calibration methods except methods
#3, #8 and #9. In these cases only one standard was used for
calibration. Source PDF was used with calibration methods #1, #4, #6
and #8. By comparing the average results and the standard deviations
obtained for method #1 to #2, #4 to #5, #6 to #7, and #8 to #9 in
Table 4.8.1., it can be seen that there is no significant difference
in the results; therefore, source or
4.8.3. An internal standard was present in all of the samples used but results were calculated both with the internal standard correction and without it for calibration methods #1 through #5. (Tables 4.8.1. to 4.8.9.). For all of the analyses, automatic liquid sampling devices were used with a single injection of each sample. At the bottom of Table 4.8.1. are the average results for all the PDFs using all the calibration methods calculated with both the internal standard (Istd) and external standard (Estd) procedures. From this data there appears to be no real difference between the results using the Istd correction and not (Estd). The use of an internal standard is left to the judgment of the analyst since the lengthening of the analysis and possible interferences caused by an internal standard compound will be different for each set of samples.
4.8.4. Three different techniques of setting the baseline during analysis were investigated. One technique was to allow the data system (Hewlett-Packard 3357) to calculate the baseline and set it automatically. The other techniques require the analyst to control the baseline by using either a basic program to set the baseline and integrate the area under the chromatogram or an "integrate function" built into the data system to set the baseline.
- 4.8.4.1. At lower concentrations of PDFs, the technique of
allowing the data system to automatically set the baseline
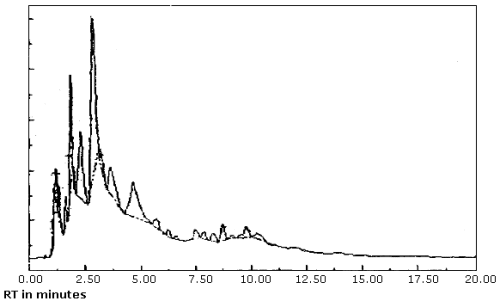
produced inconsistent results. (Figure 4.8.1. and 4.8.2.) This may
be due to a parameter in the data system termed "slope
sensitivity", but since single analytes are often requested in
addition to PDF, setting the slope sensitivity for PDF may not be
accurate for the single analytes. Calibration methods #6, #7, #8
and #9 used this technique (Tables 4.8.1. - 4.8.9.). The results
in Table 4.8.1. are the average recoveries for each calibration
technique with the 8 different PDFs. As can be seen in this table,
the percent recoveries for each separate PDF using calibration
methods #6, #7, #8 and #9 ranged from
4.8.4.2. Calibration methods #1 and #2 used a basic program for baseline setting and integration. This basic program was written to be used after analyzing the standards, blanks and samples. The raw data collected during an analysis is in the form of area slices which are simply detector voltages taken and stored every 0.5 s. The analyst enters into the basic program the time span over which the PDF constituents elute. The program saves the value of the first area slice in the analytical run to be used as the baseline and when the start time of the PDF is reached the program subtracts the baseline area slice from all the area slices in the specified time span and sums the differences. This summation is used as the area of PDF constituents. This program integrated the area above the baseline but not as individual peaks. The average recoveries are presented in Table 4.8.1. Since this program did not have any peak detection routine, it would not differentiate between a rise in the baseline due to a peak and column bleed. Therefore, if the baseline was not consistent and PDF constituents were eluting from the column at these times, area may be added to the PDF area which was caused by column bleed and not PDF constituents. This technique of baseline control is not recommended.
4.8.4.3. The two evaluated integrate functions which control the baseline were "hold the baseline" (Figure. 4.8.2.) and "valley reset" (Figure 4.8.4.). The "valley reset" function resets the baseline every time the data system detects a zero slope or a switch from negative to positive slope of the detector output. This function is performed by the data system with start and stop times entered by the analyst. Calibration method #3 used this function and the area of the largest peak for calibration of a response factor. As can be seen in Table 4.8.1., the average results for all the PDFs analyzed with method #4 were 102(±2.3)% with the internal standard procedure and 102(±4.1)% with the external standard procedure. Comparing these results to those of the other calibration methods, method #4 is the most accurate. However, this method requires that the source PDF be used as analytical standards because the ratio of the area of the chosen peak to the others in the PDF must be constant.
4.8.4.4. The "hold the baseline" function simply records the detector voltage at a certain time during the analysis and maintains that as the baseline until the function is canceled. The time to start this function is slightly before the PDF constituents begin to elute and the time to cancel it is after the constituents have eluted or when column bleed becomes significant. Both of these times are set by the analyst. After the function is canceled, the data system is free to set the baseline and it usually does correct for baseline drift due to column bleed; therefore, excess area is not added to the PDF as it was with the basic program. Calibration methods #4 and 5 used this technique. The average results and standard deviations for all PDFs for these two methods given at the bottom of Table 4.8.1. are better than the other calibration methods except #3, although this calibration method (#3) requires the use of source PDF in preparing analytical standards. Therefore, using the integrate function of "hold the baseline" is recommended and a linear least squares fit of the standards should be used to quantitate the samples.
4.8.5. Recommendations
For analysis of petroleum distillate fractions, either the source
PDF (Section 3.3.2.) or a
Average Percent Recoveries
Calculated from Tables 4.8.2. to 4.8.9.
|
| ||||||||||
| (see notes) | calibration methods | |||||||||
| table | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| ||||||||||
| 4.8.2. | yes | 105 | 96 | 104 | 107 | 95 | 97 | 92 | 100 | 93 |
| no | 103 | 95 | 100 | 102 | 95 | x | x | x | x | |
| 4.8.3. | yes | 106 | 115 | 104 | 100 | 111 | 99 | 101 | 110 | 110 |
| no | 108 | 115 | 104 | 106 | 109 | x | x | x | x | |
| 4.8.4. | yes | 109 | 104 | 99 | 91 | 99 | 93 | 113 | 91 | 93 |
| no | 115 | 106 | 103 | 94 | 98 | x | x | x | x | |
| 4.8.5. | yes | 103 | 102 | 104 | 90 | 83 | 110 | 93 | 93 | 91 |
| no | 103 | 105 | 102 | 87 | 83 | x | x | x | x | |
| 4.8.6. | yes | 99 | 97 | 100 | 104 | 103 | 95 | 84 | 75 | 75 |
| no | 98 | 96 | 99 | 103 | 103 | x | x | x | x | |
| 4.8.7. | yes | 100 | 95 | 104 | 103 | 104 | 107 | 110 | 31 | 32 |
| no | 99 | 97 | 100 | 100 | 102 | x | x | x | x | |
| 4.8.8. | yes | 95 | 91 | 100 | 106 | 99 | 143 | 100 | 29 | 28 |
| no | 104 | 93 | 109 | 114 | 101 | x | x | x | x | |
| 4.8.9. | yes | 119 | 125 | 100 | 99 | 100 | 83 | 73 | 67 | 73 |
| no | 135 | 135 | 95 | 95 | 95 | x | x | x | x | |
| 105 | 103 | 102 | 100 | 99 | 103 | 96 | 74 | 74 | ||
| SD | 7.3 | 11.5 | 2.3 | 6.4 | 8.1 | 18.0 | 13.2 | 30.6 | 29.7 | |
| 108 | 105 | 102 | 100 | 98 | x | x | x | x | ||
| SD | 12.1 | 14.1 | 4.1 | 8.2 | 7.7 | x | x | x | x | |
|
| ||||||||||
| notes: 1.) Explanation of Calibration methods under table 4.8.2. 2.) Istd column: "yes" indicates internal standard was used; "no" indicates an external standard procedure used. 3.) "x" under calibration methods #6, 7, 8 9 indicates no data was collected with an external standard procedure. | ||||||||||
Percent Found for Stoddard solvent A
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 1 | 789 | yes | 104 | 96 | 102 | 111 | 98 | 96 | 91 | 101 | 93 |
| no | 102 | 93 | 97 | 104 | 96 | x | x | x | x | ||
| 2 | 3159 | yes | 101 | 94 | 103 | 106 | 94 | 99 | 93 | 102 | 94 |
| no | 100 | 92 | 98 | 100 | 93 | x | x | x | x | ||
| 3 | 4739 | yes | 102 | 94 | 104 | 107 | 95 | 99 | 92 | 101 | 93 |
| no | 101 | 94 | 100 | 103 | 95 | x | x | x | x | ||
| 4 | 237 | yes | 120 | 103 | 107 | 109 | 97 | 91 | 87 | 96 | 88 |
| no | 108 | 98 | 102 | 101 | 94 | x | x | x | x | ||
| 5 | 6318 | yes | 103 | 94 | 104 | 104 | 93 | 103 | 96 | 104 | 96 |
| no | 101 | 94 | 101 | 101 | 93 | x | x | x | x | ||
| 6 | 3159 | yes | 102 | 95 | 105 | 105 | 94 | 102 | 102 | 105 | 9 |
| no | 103 | 95 | 101 | 101 | 94 | x | x | x | x | ||
| 7 | 6318 | yes | 103 | 94 | 104 | 106 | 94 | 101 | 93 | 102 | 94 |
| no | 101 | 95 | 101 | 103 | 95 | x | x | x | x | ||
| 8 | 789 | yes | 102 | 94 | 101 | 107 | 95 | 91 | 86 | 95 | 88 |
| no | 100 | 92 | 97 | 102 | 95 | x | x | x | x | ||
| 9 | 4739 | yes | 103 | 95 | 105 | 107 | 95 | 102 | 95 | 104 | 96 |
| no | 103 | 95 | 102 | 104 | 96 | x | x | x | x | ||
| 10 | 2369 | yes | 102 | 95 | 104 | 108 | 96 | 97 | 92 | 101 | 93 |
| no | 103 | 95 | 101 | 104 | 97 | x | x | x | x | ||
| 11 | 237 | yes | 115 | 99 | 105 | 104 | 92 | 86 | 81 | 90 | 83 |
| no | 105 | 95 | 101 | 99 | 91 | x | x | x | x | ||
| 12 | 2369 | yes | 104 | 97 | 106 | 110 | 98 | 99 | 94 | 97 | 95 |
| no | 106 | 97 | 104 | 107 | 99 | x | x | x | x | ||
|
| |||||||||||
| notes: 1.) Calibration method #1 uses as analytical standards the source PDF, the basic program for peak integration and area summation of the standards for calibration. 2.) Calibration method #2 uses as analytical standards a 3.) Calibration method #3 uses the source PDF, "valley reset" for peak integration and a single peak in the standards for calibration. 4.) Calibration method #4 uses as analytical standards the source PDF, "hold the baseline" for peak integration and area summation of standards for calibration. 5.) Calibration method #5 uses as analytical standards a 6.) Calibration method #6 uses as analytical standards the source PDF, the data system sets the baseline for peak integration, and area summation of standards for calibration. 7.) Calibration method #7 uses as analytical standards a 8.) Calibration method #8 uses as analytical standards the source PDF, the data system sets the baseline for peak integration, and area summation of only one standard for calibration. 9.) Calibration method #9 uses as analytical standards a | |||||||||||
Percent Found for Stoddard Solvent B
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 13 | 3109 | yes | 112 | 119 | 111 | 116 | 128 | 103 | 95 | 103 | 103 |
| no | 107 | 114 | 106 | 111 | 118 | x | x | x | x | ||
| 14 | 777 | yes | 111 | 120 | 108 | 104 | 120 | 125 | 122 | 137 | 136 |
| no | 108 | 116 | 103 | 100 | 111 | x | x | x | x | ||
| 15 | 233 | yes | 122 | 141 | 103 | 89 | 96 | 79 | 132 | 136 | 136 |
| no | 117 | 125 | 94 | lost | 89 | x | x | x | x | ||
| 16 | 5440 | yes | 106 | 113 | 106 | 106 | 117 | 107 | 98 | 105 | 105 |
| no | 104 | 110 | 104 | 104 | 112 | x | x | x | x | ||
| 17 | 7772 | yes | 106 | 114 | 104 | 105 | 116 | 107 | 103 | 106 | 105 |
| no | 104 | 110 | 103 | 105 | 112 | x | x | x | x | ||
| 18 | 233 | yes | 107 | 125 | 103 | 79 | 78 | 55 | 101 | 114 | 113 |
| no | 108 | 116 | 103 | lost | 76 | x | x | x | x | ||
| 19 | 4663 | yes | 101 | 108 | 101 | lost | 113 | 99 | 89 | 98 | 98 |
| no | 107 | 114 | 106 | 107 | 115 | x | x | x | x | ||
| 20 | 3109 | yes | 100 | 106 | 100 | 99 | 114 | 97 | 86 | 97 | 97 |
| no | 109 | 116 | 107 | 106 | 119 | x | x | x | x | ||
| 21 | 777 | yes | 99 | 108 | 100 | 97 | 109 | 105 | 102 | 118 | 118 |
| no | 104 | 112 | 103 | 100 | 109 | x | x | x | x | ||
| 22 | 7772 | yes | 104 | 112 | 103 | 104 | 114 | 105 | 101 | 104 | 104 |
| no | 106 | 113 | 107 | 108 | 115 | x | x | x | x | ||
| 23 | 5440 | yes | 103 | 110 | 104 | 104 | 115 | 104 | 95 | 103 | 103 |
| no | 110 | 117 | 111 | 111 | 119 | x | x | x | x | ||
| 24 | 4663 | yes | 100 | 107 | 101 | 102 | 113 | 99 | 89 | 98 | 98 |
| no | 107 | 114 | 108 | 108 | 116 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.2. | |||||||||||
Percent Found for V.M.&P. Naphtha A
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 25 | 7528 | yes | 103 | 102 | 104 | 89 | 98 | 102 | 104 | 102 | 104 |
| no | 120 | 105 | 106 | 94 | 98 | x | x | x | x | ||
| 26 | 5270 | yes | 102 | 104 | 103 | 89 | 97 | 101 | 105 | 102 | 104 |
| no | 112 | 107 | 107 | 95 | 99 | x | x | x | x | ||
| 27 | 7528 | yes | 106 | 104 | 107 | 92 | 100 | 105 | 107 | 105 | 107 |
| no | 119 | 105 | 106 | 94 | 98 | x | x | x | x | ||
| 28 | 1506 | yes | 106 | 107 | 98 | 92 | 100 | 93 | 105 | 93 | 95 |
| no | 110 | 109 | 105 | 98 | 102 | x | x | x | x | ||
| 29 | 3011 | yes | 100 | 103 | 97 | 88 | 96 | 98 | 104 | 98 | 100 |
| no | 106 | 106 | 104 | 94 | 98 | x | x | x | x | ||
| 30 | 226 | yes | 172 | 119 | 96 | 100 | 110 | 72 | 148 | 65 | 66 |
| no | 177 | 121 | 101 | 100 | 102 | x | x | x | x | ||
| 31 | 753 | yes | 98 | 99 | 94 | 85 | 93 | 88 | 111 | 86 | 88 |
| no | 99 | 99 | 99 | 88 | 92 | x | x | x | x | ||
| 32 | 5270 | yes | 99 | 102 | 101 | 88 | 96 | 101 | 103 | 100 | 102 |
| no | 106 | 103 | 103 | 92 | 96 | x | x | x | x | ||
| 33 | 753 | yes | 101 | 103 | 94 | 91 | 99 | 91 | 114 | 89 | 91 |
| no | 101 | 102 | 98 | 92 | 96 | x | x | x | x | ||
| 34 | 1506 | yes | 100 | 106 | 98 | 92 | 100 | 93 | 105 | 93 | 95 |
| no | 103 | 108 | 105 | 97 | 101 | x | x | x | x | ||
| 35 | 226 | yes | 124 | 103 | 95 | 97 | 106 | 71 | 146 | 64 | 65 |
| no | 126 | 103 | 99 | 93 | 96 | x | x | x | x | ||
| 36 | 3011 | yes | 97 | 103 | 98 | 89 | 97 | 98 | 104 | 98 | 100 |
| no | 103 | 106 | 105 | 95 | 99 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.2. | |||||||||||
Percent Found for V.M.&P. Naphtha B
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 37 | 3768 | yes | 103 | 98 | 106 | 96 | 88 | 103 | 98 | 101 | 99 |
| no | 95 | 93 | 97 | 86 | 83 | x | x | x | x | ||
| 38 | 6029 | yes | 102 | 100 | 110 | 96 | 87 | 103 | 99 | 103 | 101 |
| no | 95 | 98 | 97 | 86 | 82 | x | x | x | x | ||
| 39 | 754 | yes | 102 | 100 | 101 | 87 | 80 | 106 | 84 | 87 | 85 |
| no | 94 | 94 | 93 | 73 | 70 | x | x | x | x | ||
| 40 | 2261 | yes | 106 | 100 | 105 | 97 | 89 | 100 | 92 | 95 | 93 |
| no | 99 | 95 | 98 | 88 | 85 | x | x | x | x | ||
| 41 | 301 | yes | 95 | 109 | 100 | 72 | 66 | 111 | 54 | 58 | 57 |
| no | 90 | 106 | 94 | 52 | 50 | x | x | x | x | ||
| 42 | 4522 | yes | 101 | 97 | 102 | 92 | 85 | 100 | 97 | 100 | 98 |
| no | 104 | 105 | 104 | 94 | 90 | x | x | x | x | ||
| 43 | 3768 | yes | 104 | 99 | 105 | 94 | 86 | 104 | 99 | 102 | 100 |
| no | 107 | 106 | 107 | 96 | 86 | x | x | x | x | ||
| 44 | 2261 | yes | 106 | 99 | 104 | 95 | 87 | 102 | 95 | 97 | 95 |
| no | 109 | 104 | 108 | 98 | 94 | x | x | x | x | ||
| 45 | 301 | yes | 113 | 124 | 101 | 77 | 70 | 127 | 70 | 74 | 73 |
| no | 117 | 129 | 105 | 79 | 75 | x | x | x | x | ||
| 46 | 6028 | yes | 102 | 100 | 111 | 95 | 87 | 103 | 100 | 103 | 101 |
| no | 107 | 114 | 110 | 98 | 94 | x | x | x | x | ||
| 47 | 754 | yes | 106 | 104 | 191 | 87 | 81 | 157 | 133 | 89 | 87 |
| no | 113 | 111 | 108 | 95 | 91 | x | x | x | x | ||
| 48 | 4522 | yes | 103 | 97 | 106 | 94 | 86 | 103 | 99 | 102 | 100 |
| no | 109 | 111 | 112 | 100 | 95 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.2. | |||||||||||
Percent Found for Stoddard Solvent D
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 49 | 3897 | yes | 99 | 99 | 101 | 100 | 98 | 100 | 90 | 88 | 88 |
| no | 98 | 97 | 98 | 98 | 97 | x | x | x | x | ||
| 50 | 6235 | yes | 99 | 98 | 101 | 98 | 98 | 94 | 88 | 88 | 88 |
| no | 97 | 96 | 99 | 97 | 97 | x | x | x | x | ||
| 51 | 779 | yes | 96 | 92 | 97 | 106 | 106 | 96 | 78 | 61 | 61 |
| no | 95 | 91 | 96 | 106 | 105 | x | x | x | x | ||
| 52 | 545 | yes | 92 | 87 | 95 | 105 | 105 | 105 | 82 | 59 | 59 |
| no | 91 | 85 | 94 | 104 | 104 | x | x | x | x | ||
| 53 | 6235 | yes | 100 | 99 | 102 | 99 | 98 | 95 | 88 | 89 | 88 |
| no | 100 | 99 | 102 | 99 | 99 | x | x | x | x | ||
| 54 | 2338 | yes | 102 | 101 | 102 | 106 | 105 | 109 | 95 | 89 | 89 |
| no | 100 | 99 | 100 | 104 | 104 | x | x | x | x | ||
| 55 | 545 | yes | 99 | 94 | 101 | 112 | 112 | 69 | 82 | 60 | 60 |
| no | 98 | 93 | 100 | 112 | 112 | x | x | x | x | ||
| 56 | 3897 | yes | 101 | 100 | 102 | 101 | 100 | 101 | 91 | 89 | 89 |
| no | 100 | 100 | 101 | 100 | 100 | x | x | x | x | ||
| 57 | 1559 | yes | 100 | 99 | 101 | 105 | 105 | 94 | 79 | 70 | 70 |
| no | 101 | 99 | 101 | 106 | 105 | x | x | x | x | ||
| 58 | 2338 | yes | 101 | 100 | 101 | 103 | 102 | 89 | 77 | 71 | 71 |
| no | 100 | 99 | 100 | 101 | 101 | x | x | x | x | ||
| 59 | 1559 | yes | 100 | 98 | 101 | 105 | 104 | 93 | 79 | 70 | 70 |
| no | 102 | 100 | 102 | 107 | 106 | x | x | x | x | ||
| 60 | 779 | yes | 100 | 96 | 100 | 105 | 105 | 99 | 80 | 63 | 63 |
| no | 767 | 739 | 769 | 810 | 809 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.2. | |||||||||||
Percent Found for Stoddard Solvent D
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 61 | 3045 | yes | 102 | 100 | 102 | 103 | 102 | 102 | 104 | 34 | 34 |
| no | 96 | 100 | 96 | 98 | 103 | x | x | x | x | ||
| 62 | 3045 | yes | 102 | 101 | 102 | 104 | 103 | 102 | 104 | 34 | 34 |
| no | 96 | 101 | 97 | 98 | 103 | x | x | x | x | ||
| 63 | 6853 | yes | 103 | 102 | 104 | 102 | 102 | 103 | 105 | 34 | 34 |
| no | 100 | 101 | 99 | 98 | 102 | x | x | x | x | ||
| 64 | 1523 | yes | 98 | 94 | 96 | 101 | 104 | 100 | 102 | 30 | 31 |
| no | 97 | 98 | 94 | 100 | 101 | x | x | x | x | ||
| 65 | 761 | yes | 97 | 89 | 99 | 104 | 107 | 114 | 116 | 29 | 30 |
| no | 100 | 92 | 97 | 102 | 104 | x | x | x | x | ||
| 66 | 533 | yes | 99 | 87 | 119 | 107 | 110 | 125 | 127 | 28 | 28 |
| no | 106 | 90 | 117 | 105 | 106 | x | x | x | x | ||
| 67 | 6853 | yes | 99 | 97 | 100 | 98 | 101 | 98 | 100 | 33 | 33 |
| no | 98 | 99 | 97 | 96 | 97 | x | x | x | x | ||
| 68 | 533 | yes | 99 | 87 | 100 | 107 | 108 | 125 | 127 | 28 | 29 |
| no | 105 | 88 | 96 | 103 | 106 | x | x | x | x | ||
| 69 | 1523 | yes | 98 | 94 | 101 | 101 | 103 | 100 | 102 | 30 | 31 |
| no | 96 | 97 | 97 | 98 | 100 | x | x | x | x | ||
| 70 | 761 | yes | 101 | 93 | 119 | 108 | 109 | 117 | 119 | 30 | 31 |
| no | 102 | 94 | 115 | 104 | 108 | x | x | x | x | ||
| 71 | 4568 | yes | 100 | 99 | 99 | 100 | 101 | 99 | 102 | 33 | 34 |
| no | 9699 | 95 | 96 | 99 | x | x | x | x | |||
| 72 | 4568 | yes | 100 | 98 | 104 | 100 | 102 | 99 | 101 | 33 | 34 |
| no | 96 | 100 | 100 | 97 | 99 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.2. | |||||||||||
Percent Found for Stoddard solvent E
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 73 | 7756 | yes | 104 | 94 | 103 | 99 | 92 | 106 | 102 | 35 | 34 |
| no | 108 | 96 | 111 | 106 | 94 | x | x | x | x | ||
| 74 | 2327 | yes | 103 | 98 | 103 | 103 | 95 | 153 | 105 | 35 | 34 |
| no | 110 | 100 | 112 | 109 | 97 | x | x | x | x | ||
| 75 | 3878 | yes | 104 | 97 | 102 | 100 | 93 | 132 | 102 | 35 | 34 |
| no | 110 | 98 | 111 | 106 | 95 | x | x | x | x | ||
| 76 | 776 | yes | 89 | 88 | 96 | 116 | 108 | 139 | 77 | 17 | 16 |
| no | 99 | 88 | 103 | 122 | 109 | x | x | x | x | ||
| 77 | 5429 | yes | 101 | 94 | 102 | 97 | 90 | 116 | 98 | 34 | 32 |
| no | 108 | 96 | 109 | 104 | 93 | x | x | x | x | ||
| 78 | 7756 | yes | 102 | 93 | 101 | 96 | 89 | 103 | 97 | 33 | 32 |
| no | 110 | 97 | 112 | 106 | 94 | x | x | x | x | ||
| 79 | 388 | yes | 78 | 81 | 99 | 130 | 125 | 206 | 112 | 17 | 16 |
| no | 91 | 80 | 106 | 140 | 126 | x | x | x | x | ||
| 80 | 3878 | yes | 101 | 94 | 103 | 98 | 91 | 129 | 99 | 34 | 33 |
| no | 108 | 97 | 110 | 105 | 94 | x | x | x | x | ||
| 81 | 5429 | yes | 102 | 94 | 103 | 99 | 92 | 118 | 100 | 35 | 33 |
| no | 111 | 99 | 112 | 109 | 97 | x | x | x | x | ||
| 82 | 2327 | yes | 100 | 96 | 102 | 101 | 94 | 151 | 109 | 34 | 33 |
| no | 110 | 99 | 112 | 108 | 97 | x | x | x | x | ||
| 83 | 776 | yes | 84 | 83 | 95 | 110 | 102 | 170 | 97 | 24 | 23 |
| no | 96 | 86 | 104 | 117 | 105 | x | x | x | x | ||
| 84 | 388 | yes | 77 | 79 | 98 | 122 | 114 | 199 | 108 | 16 | 15 |
| no | 92 | 80 | 107 | 132 | 118 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.2. | |||||||||||
Results for Mineral Spirits A
|
| |||||||||||
| (see notes) | calibration methods | ||||||||||
| sample | µg | Istd | #1 | #2 | #3 | #4 | #5 | #6 | #7 | #8 | #9 |
|
| |||||||||||
| 85 | 7673 | yes | 109 | 113 | 106 | 101 | 106 | 103 | 99 | 94 | 100 |
| no | 100 | 98 | 88 | 91 | 90 | x | x | x | x | ||
| 86 | 230 | yes | 186 | 200 | 108 | 90 | 88 | 57 | 109 | 43 | 46 |
| no | 270 | 275 | 98 | 82 | 94 | x | x | x | x | ||
| 87 | 1534 | yes | 149 | 158 | 119 | 129 | 135 | 110 | 93 | 86 | 92 |
| no | 144 | 145 | 107 | 119 | 117 | x | x | x | x | ||
| 88 | 5371 | yes | 107 | 110 | 103 | 102 | 107 | 100 | 92 | 86 | 92 |
| no | 115 | 113 | 106 | 108 | 107 | x | x | x | x | ||
| 89 | 7673 | yes | 106 | 110 | 103 | 96 | 101 | 107 | 104 | 99 | 106 |
| no | 116 | 113 | 107 | 102 | 101 | x | x | x | x | ||
| 90 | 537 | yes | 210 | 224 | 65 | 123 | 114 | 50 | 40 | 37 | 40 |
| no | 226 | 228 | 67 | 108 | 106 | x | x | x | x | ||
| 91 | 2302 | yes | 110 | 115 | 104 | 104 | 107 | 89 | 76 | 70 | 75 |
| no | 112 | 112 | 101 | 102 | 99 | x | x | x | x | ||
| 92 | 1534 | yes | 107 | 113 | 106 | 106 | 108 | 91 | 76 | 70 | 75 |
| no | 112 | 112 | 103 | 103 | 102 | x | x | x | x | ||
| 93 | 537 | yes | 61 | 65 | 71 | 62 | 56 | 39 | 32 | 30 | 31 |
| no | 73 | 74 | 64 | 54 | 54 | x | x | x | x | ||
| 94 | 230 | yes | 82 | 89 | 106 | 72 | 78 | 45 | 36 | 33 | 35 |
| no | 143 | 149 | 96 | 67 | 66 | x | x | x | x | ||
| 95 | 5371 | yes | 99 | 103 | 101 | 93 | 97 | 110 | 101 | 95 | 102 |
| no | 106 | 105 | 106 | 99 | 97 | x | x | x | x | ||
| 96 | 2302 | yes | 104 | 110 | 106 | 106 | 110 | 90 | 77 | 71 | 76 |
| no | 103 | 103 | 104 | 104 | 102 | x | x | x | x | ||
|
| |||||||||||
| note: Explanation of calibration methods under Table 4.8.1. | |||||||||||
5. References
- 5.1. "Criteria for a Recommended Standard...Occupational Exposure
to Refined Petroleum Solvents"; Department of Health, Education and
Welfare, National Institute for Occupational Safety and Health:
Cincinnati, OH, 1977 (DHEW) (NIOSH) Publ. (U.S.) No. 77-192.
5.2. "NIOSH Manual of Analytical Methods", 2nd ed.; Department of Health, Education and Welfare, National Institute for Occupational Safety and Health: Cincinnati, OH, 1977; Vol. 3, Methods S380 and S382; DHEW (NIOSH) Publ. (U.S.) No. 77-157-C.
5.3. Drushel, Harry V. Journal of Chromatographic Science. 21, August 1983, p 375.
5.4. "Occupational Health Guideline for Stoddard Solvent", Department of Health and Human Services, National Institute for Occupational Safety and Health: U.S. Government Printing Office, Washington, D.C., 1978; Publ. 81-123.
5.5. "Occupational Health Guideline for Petroleum Distillates", Department of Health and Human Services, National Institute for Occupational Safety and Health: U.S. Government Printing Office, Washington, D.C. 1978; Publ. 81-123.