HEXAVALENT CHROMIUM IN WORKPLACE ATMOSPHERES
| OSHA Method Number: | ID-215 (This method supersedes ID-103) |
| Matrix: | Air |
| OSHA Permissible Exposure Limit (proposed) Hexavalent Chromium [Cr(VI)] Time Weighted Average (TWA): Action Level (AL): |
0.50
µg/m3 0.25 µg/m3 |
| Collection Device: | An air sample is collected using a |
| Recommended Sampling Rate: | 2 liters per minute (L/min) |
| Recommended Air Volume: TWA and AL: |
960 L (2 L/min for 480 min) |
| Analytical Procedure: | The hexavalent chromium, Cr(VI), is extracted from the PVC
filter using an aqueous solution containing 10% sodium carbonate
Na2CO3)/ 2%
sodium bicarbonate (NaHCO3) and the
mixture of phosphate buffer/magnesium sulfate [~10 mg as Mg (II)].
After dilution, an aliquot of this solution is analyzed for Cr(VI)
by an ion chromatograph equipped with a |
| Detection Limit Qualitative: Quantitative: |
1.0 × 10-3
µg/m3 as Cr(VI) (960-L air
sample) 3.0 × 10-3 µg/m3 as Cr(VI) (960-L air sample) |
| Precision and Accuracy (Soluble and Insoluble) Validation Range: CV1(pooled): Bias: Overall Error: |
0.12 to 0.42 µg/m3
(960-L air sample) 0.059 -0.004 ±12.9% |
| Method Classification: | Validated Method |
| Chemists: | James C. Ku, Mary Eide |
| Date: | June, 1998 |
Commercial manufacturers and products mentioned in this method are
for descriptive use only and do not constitute endorsements byUSDOL-OSHA.
Similar products from other sources can be substituted.
Branch of Inorganic Methods Development
OSHA Salt Lake
Technical Center
Salt Lake City, Utah
1. Introduction
This method describes the sample collection and analysis of airborne
hexavalent chromium, Cr(VI). This method should be used by industrial
hygienists experienced in monitoring for exposures and analysts
experienced in the use of ion chromatography and the interpretation of ion
chromatograms. Samples are taken in the breathing zone of workplace
personnel, and analysis is performed with an ion chromatograph (IC)
equipped with a
1.1. History
To sample for Cr(VI) in the workplace, a 37-mm diameter,
Recently, a reduction in the Permissible Exposure Limit (PEL) for
Cr(VI) has been proposed by OSHA, with 0.50
µg/m3 for the Time Weighted Average
(TWA) and 0.25 µg/m3 for the Action
Level (AL). The differential pulse polarographic method was not
sufficiently sensitive to quantitate at the proposed levels, and a new
method was developed using an IC equipped with a
1.2. Principle
Hexavalent chromium is collected on a 37-mm diameter PVC filter. Any compound existing in the Cr(VI) valence state is extracted from the PVC filter using a hot aqueous solution containing 10% sodium carbonate (Na2CO3), 2% sodium bicarbonate (NaHCO3), and the phosphate buffer/magnesium sulfate mixture. The reaction between any chromate species and carbonate is illustrated by the following equation (5.3.):
MCrO4 + CO32- ------› MCO3 + CrO42-
Where M = metals (e.g., lead, zinc, cadmium, sodium, potassium,
calcium, etc.)
In the presence of a large excess of carbonate, the
equilibrium is shifted quantitatively to the right. Any chromate
compounds (soluble and insoluble) contained in the sample are
converted to their corresponding soluble carbonates. Interferences are
minimized by the addition of the magnesium.
After dilution, an aliquot of this extract is analyzed for Cr(VI)
with an IC equipped with a postcolumn reagent delivery module and a
1.3. Advantages and Disadvantages
1.3.1. This method has adequate sensitivity for determining compliance with the proposed OSHA TWA and AL PELs for Cr(VI) exposure.
1.3.2. The method is simple, rapid, and easily automated.
1.3.3. The method is specific and can determine Cr(VI) in the presence of Cr(III). Most heavy metals, such as vanadium, copper, iron (III), and molybdenum, do not significantly interfere. Fe(II) appears to cause a negative interference during sampling and storage (see Sections 1.5. and 4.4. for further information).
1.3.4. By using alkaline extraction conditions (pH = 10 to 11) in
which Cr(VI) is more stable, sample recovery is improved by
preventing Cr(VI) losses which may occur in a more acidic extraction
media. Both water soluble and insoluble Cr(VI) compounds are soluble
in the alkaline
1.3.5. Extraction and preparation of samples for analyses involve simple procedures and equipment.
1.3.6. If necessary, the amount of Cr(VI) can also be analyzed
and confirmed by differential pulse polarography (DPP), provided
samples and standards are
1.3.7. A disadvantage is the extraction solution and sulfuric
acid used are very caustic. The extraction solution may also limit
the column life and type of instrumentation used. The module used in
this method is equipped with a reagent reservoir, a mixing
tee/reaction coil system, and a
1.4. Method Performance
A synopsis of the method performance is presented below. Further information can be found in Section 4.
1.4.1. This method was validated using soluble and insoluble
chromate compounds. The compounds used were potassium dichromate and
lead chromate for soluble and insoluble chromate, respectively. The
significant availability and industrial use of potassium dichromate
indicated it was a goodchoice to represent the chemical
characteristics of the soluable chromates for this evaluation.
Solubility product values indicated that lead chromate was the least
soluble of the chromate compounds commonly found in industry,
therefore it was chosen to represent the insoluble chromate. Filter
samples were spiked with about 0.11 to 0.40 µg [as Cr(VI)].
Using an
1.4.2. The qualitative detection limit was 0.001 µg as
Cr(VI) when using a
1.4.3. The quantitative detection limit was 0.003 µg as
Cr(VI) when using a
1.4.4. The sensitivity of the analytical method, when using the
instrumental parameters listed in Section 3.6., was calculated from
the slope of a linear working range curve [0.5 to 1,000 ng/mL
Cr(VI)]. The sensitivity was 2.47 × 104
area units per 1 ng/mL, when using a Dionex Series 4500i ion
chromatograph with AI450 computer software (Dionex, Sunnyvale, CA).
The sensitivity was 1.57 × 104 area units
per 1 ng/mL, when using a Dionex DX500 ion chromatograph with a 10
mm cell and a 150 µL sample loop (Dionex, Sunnyvale, CA). The
sensitivity of this method was significantly better than OSHA Method
No.
1.4.5. The total pooled coefficients of variation (CV1), bias, and total overall error (OE) are as follows:
For soluble chromate:
CV1
(pooled) = 0.054; bias
= + 0.007;
OET = ±11.5%
For insoluble chromate:
CV1
(pooled) = 0.064; bias
= - 0.014;
OET = ±14.2%
For both types of chromate compounds (pooled soluble and
insoluble):
CV1 (pooled)
= 0.059; bias = -
0.004; OET
= ±12.9%
1.4.6. The collection efficiency of 0.945 ± 0.035 has been previous determined for chromic acid mist collected on PVC filters (5.11.).
1.4.7. Quality control (QC) samples were prepared as single blind
samples by spiking aqueous solutions of potassium dichromate on PVC
filters. Amounts spiked ranged from 10 to 20 µg. Results of
samples analyzed from 1982-89 using the DPP technique, and samples
analyzed using this method (IC/
| DPP* | IC/UV-vis | |
| Samples (N): | 282 | 57 |
| Average recovery: | 94.1% | 94.8% |
| CV1(pooled): | 0.10 | 0.054 |
| *DPP data obtained from reference 5.12. | ||
1.4.8. Samples can be stored at ambient (20 to 25 °C) temperature on a lab bench for a period of at least 30 days. The mean sample recovery after 30 days of storage was within ±5% of the recovery at Day 0.
1.5. Interferences
1.5.1. Reducing species such as Cr(III), V(III), and Cu(I), etc.
in
1.5.2. A positive interference can be any substance that has the same retention time as Cr(VI), and absorbs light at 540 nm wavelength when using the ion chromatographic operating conditions described in this method. Changing the chromatographic separation conditions (detector settings, column, eluent flow rate, and strength, etc.) may minimize the interference. None of the more common metallic species coexisting with Cr(VI) in the workplace and potentially soluble in the extraction solution were found to positively interfere when using the analytical conditions described in this method. A positive interference from Cr(III) can occur when extracted with BE or a more strongly basic extraction solution for spray paint samples (SPE) alone; however, the addition of the phosphate buffer/Mg(II) solution to the extraction process minimizes this positive interference. For samples having Cr(III) levels of 1 µg/mL, the positive interference changed from <1% for BE to <0.02% for BE with phosphate buffer/Mg(II). For SPE samples containing 10 µg/mL Cr(III), the positive interference changed from <0.2% for SPE to <0.03% for SPE with phosphate buffer/Mg(II) (see Sections 4.4.1, 4.4.3, and 4.4.4).
1.6. Uses
The principal commercial Cr(VI) compounds are chromium trioxide (chromic acid anhydride), and the chromates and dichromates of sodium, potassium, ammonium, calcium, barium, zinc, strontium, and lead. They are used as oxidizing agents in tanning, photography, dyeing, and electroplating, and as rust inhibitors and pigments.
1.7. Physical and Chemical Properties of Certain Chromates (5.15.)
| Chromium (VI) trioxide | Potassium chromate | Lead chromate | Zinc chromate | Potassium dichromate | |
| CAS No. | 1333-82-0 | 7789-00-6 | 7758-97-6 | 13530-65-9 | 7778-50-9 |
| Synonyms | Chromic acid, chromic anhydride; Chromia; Chromic trioxide | Chromic acid, dipotassium salt; Dipotassium monochromate | Chromic acid, lead salt; Crocoite; Phoenicochroite; Plumbous chromate | Chromic acid, zinc salt; Zinc tetraoxychromate; Zinc chromium oxide | Potassium bichromate; red potassium chromate |
| Description | Dark,
|
Rhombic, yellow crystals | Yellow crystals | Lemon-yellow prisms | Yellow-red crystals |
| Formula | CrO3 | K2CrO4 | PbCrO4 | ZnCrO4* | K2Cr2O7 |
| Constants and Solubility |
Mol wt: 100.01 mp: 196°C d: 2.70 Very sol in water (625 g/L at 20°C), insol in alcohol. |
Mol wt: 194.20 mp: 971°C d: 2.732 at 18°C Sol in water (1,020 g/L at 100°C), insol in alcohol. |
Mol wt: 323.22 mp: 844°C bp: decomposes d: 6.3 Very slightly sol in water (0.058 mg/L at 25°C), sol in strong acids and alkalies. |
Mol wt: 181.4 mp: not available d: 3.40 Slightly sol in water, sol in acids. |
Mol wt:294.2 mp: 396°C decomposition pt: 500°C d: 2.676 Sol in water (1.020 g/L @ 100°C) insol in alcohol |
| Fire and explosion hazard | Dangerous: a very powerful oxidizing agent. In contact with organic matter or reducing agents in general it can cause violent reactions. Upon intimate contact with powerful reducing agents it can cause violent explosions. | Moderate, by chemical reaction; a powerful oxidizer. | Moderate, by chemical reaction. | Moderate, by chemical reaction. | Moderate, by chemical reaction |
*Molecular formula was 4ZnO CrO 3 3H
2O, and confirmed
1.8. Toxicology (5.16.)
Information listed within this section is a synopsis of current knowledge of the physiological effects of chromic acid and chromates and is not intended to be used as a basis for OSHA policy.
1.8.1. Chromic acid and its salts have a corrosive action on the skin and mucous membranes. The characteristic lesion is a deep, penetrating ulcer, which, for the most part, does not tend to suppurate, and is slow in healing. Lesions are confined to the exposed area, and the skin of the nasal septum is a common site.
1.8.2. Breathing in high levels (greater than 2
µg/m3) of Cr(VI) can cause
irritation to the nasal passage, such as runny nose, sneezing,
itching, nosebleeds, ulcers, and holes in the nasal septum. These
effects have primarily occurred in factory workers who have produced
or used Cr(VI) for several months to many years.
1.8.3. Workers handling liquids or solids containing Cr(VI) compounds have developed skin ulcers.
1.8.4. Certain Cr(VI) compounds (calcium chromate, chromium trioxide, lead chromate, sodium dichromate, strontium chromate, and zinc chromate) are known animal and/or human carcinogens. The International Agency for Research on Cancer (IARC) has determined that Cr(VI) is carcinogenic to humans (Group 1), based on sufficient evidence in humans for the carcinogenicity of Cr(VI) compounds as found in chromate production, chromate pigment production, and chromium plating industries (5.17). IARC's determination is also based on sufficient evidence in experimental animals for the carcinogenicity of calcium chromate, zinc chromate, strontium chromate, and lead chromate; and limited evidence in experimental animals for the carcinogenicity of chromic acid and sodium dichromate.
2. Sampling (See Interferences, Section 1.5. before sampling.)
| Note: | Bulk samples can be collected and analyzed. Filters or wipe
samples collected on cellulose or cellulose esters are unacceptable
due to the instability of Cr(VI) on these media. Filter media used to validate this chromate method and to prepare QC samples are the PVC filters manufactured by MSA Inc. and Omega Special Instrument Co. as specified below. The Gelman GLA-5000 was also evaluated for extraction and storage and found acceptable. If a PVC filter from a different manufacturer is used, it will be necessary to at least evaluate the extraction efficiency and the storage, as it has been reported that there are interferences on some types of PVC filters which greatly reduce the hexavalent chromium to trivalent chromium. |
- Filter holder consisting of a two-piece polystyrene cassette,
37-mm diameter. - Backup pad, 37-mm, cellulose.
- Membrane filter, PVC, 37-mm, 5-µm pore size [part no.
625413, Mine Safety Appliances (MSA), Pittsburgh, PA; or cat. no.
P-503700, Omega Specialty Instrument Co., Chelmsford, MA]. - Gel bands (Omega Specialty Instrument Co., Chelmsford, MA) for sealing cassettes.
- Forceps, Teflon® coated.
2.1. Equipment
2.1.1. Calibrated personal sampling pumps capable of sampling within ±5% of the recommended flow rate of 2 L/min.
2.1.2. Tygon or other flexible tubing for connecting to pumps.
2.1.3. Plastic end plugs.
2.1.4. Sample assembly:
2.1.5. Stopwatch and bubble tube or meter for pump calibration.
2.1.6. Scintillation vials (for wipe or bulk samples),
2.2. Sampling Procedure - Air Samples
2.2.1. Place a PVC filter and a cellulose backup pad in each
2.2.2. Calibrate each personal sampling pump with a prepared
cassette
2.2.3. Attach prepared cassettes to calibrated sampling pumps
(the backup pad should face the pump) using appropriate lengths of
tubing. Place each cassette within the breathing zone on each
employee as appropriate. If possible, collect each sample for a full
work shift (approximately
2.2.4. If the filter becomes overloaded while sampling, consecutive samples using shorter sampling periods should be taken.
2.2.5. After sampling, place plastic end caps tightly on both ends of the cassette and apply OSHA Form 21 seals in such a way as to secure the end caps. Record the sampling conditions such as sampling time, air volume, etc. on the OSHA 91A form. (Note: It is very important to record the operation sampled (i.e., spray paint, chrome plating, welding, etc.).) When other compounds are known or suspected to be present in the air, record such information and transmit with the samples.
2.2.6. Use the same lots of filters and backup pads for blanks and collected samples. Handle the blank cassettes in exactly the same manner as the sample cassettes except that no air is drawn through them. Submit at least one blank cassette for each batch of ten samples.
2.3. Sampling Procedure - Wipe Samples
Wipe samples can be taken using PVC filters as the wipe media. Wear
clean, impervious, disposable gloves when taking each wipe sample. If
possible, carefully wipe a surface area covering 100
cm2. Carefully fold the wipe sample with the
exposed side in and then transfer into a
2.4. Sampling Procedure - Bulk Samples
If bulk samples are necessary, collect the bulk samples using a
grab sampling technique suitable for the particular material(s) in
use. If possible, transfer any bulk samples into
2.5. Shipment
2.5.1. Immediately send the samples to the laboratory with the OSHA 91A paperwork requesting hexavalent chromium [Cr(VI)] analysis.
2.5.2. Ship any bulk samples separately from air samples. Enclose Material Safety Data Sheets if available. Check current shipping restrictions and ship to the laboratory by the appropriate method and proper labeling.
3. Analysis
- previously cleaned with chromic acid cleaning solution,
- previously used for storage of Cr(VI) stock standard solutions, and
- previously used for storage of bulks containing high concentrations of Cr(VI).
- After the initial extraction with BE and PBM, the solutions
are transferred to
10-mL volumetric flasks. Place the sample beakers containing the remaining paint residue/filters and any blanks in an exhaust hood. - Add 1.5 mL of PBM solution and then 5 mL of SPE solution (Section 3.3.4.) to each beaker containing the filters. Swirl the beaker slowly until a white precipitate occurs. Cover the beaker with a watch glass and very slowly heat the solution on the hot plate at 135°C, with occasional swirling for 60 to 90 min. Allow extra extraction time for heavily loaded samples. DO NOT ALLOW ANY SOLUTIONS TO BOIL OR EVAPORATE TO DRYNESS. Sample loss from the conversion of Cr(VI) to Cr(III) can occur from excess heat (5.4.). Potential conversion of Cr(III) to Cr(VI) using a strong hydroxide extraction solution has also been noted (5.4.). However, the freshly precipitated magnesium hydroxide [10 mg of Mg(II)] formed suppresses the oxidation of dissolved Cr(III) to Cr(VI) (see Section 4.4. for details).
- Allow the solutions to cool to room temperature. Transfer each
solution to a
25-mL volumetric flask. Dilute to volume with DI H2O. Allow the precipitate to settle, or centrifuge to segregate the precipitate to the bottom of the sample. Alternately, cloudy samples may be filtered through a syringe prefilter. It is important that none of the precipitate is transferred into the autosampler vials, as it can clog the IC autosampler, tubing, and/or column frits.
3.1. Safety Precautions
3.1.1. Refer to appropriate IC instrument manuals,
3.1.2. Observe laboratory safety regulations and practices.
3.1.3. Certain chromate compounds have been identified as carcinogens (5.16., 5.17.). Care should be exercised when handling these compounds.
3.1.4. Some chemicals are corrosive. Use appropriate personal protective equipment such as safety glasses, goggles, face shields, gloves, and lab coat when handling corrosive chemicals.
3.1.5. The buffer/extraction (BE) and spray-paint extraction (SPE) solutions are basic and somewhat corrosive. Clean up any spills immediately. Store these solutions in polyethylene bottles. If the solutions are stored in glass, precipitated salts readily form over time from evaporation and will cause glass stoppers to seize. The strongly basic solutions will also attack the glass walls of the containers. Samples placed in glass volumetric flasks should be analyzed, properly disposed of, and the flasks rinsed and washed as soon as possible after analysis is completed and results are reported.
3.2. Equipment
3.2.1. Ion chromatograph (Model 4000i, 4500i, or DX500 Dionex, Sunnyvale, CA) equipped with a UV/vis detector and a postcolumn reagent delivery system containing a pressurized reagent reservoir with a 1-L polyethylene bottle, a post column pneumatic controller, and a mixing tee and reaction coil (Note: A membrane reactor module can be used in place of a mixing tee and reaction coil; however, extra maintenance is required, and depending on the module, additional dilution of the sample prior to analysis may be necessary.)
3.2.2. Hot plate and exhaust hood.
3.2.3. For extraction of air samples, use Phillips beakers,
borosilicate,
3.2.4. Teflon®-coated magnetic stirring bar and stirrer, or ultrasonicator.
3.2.5. Micro-analytical balance (0.01 mg).
3.2.6. Polyethylene bottles, 100-mL to 1-L size with caps with plastic liners.
3.2.7. Calibrated micro-pipettes or pipets, volumetric flasks,
beakers, and general laboratory glassware. The calibration on the
3.2.8. Automatic sampler (Dionex Model AS-1) and sample vials (0.5 mL) with filter caps.
3.2.9 . Laboratory automation system: Ion chromatograph interfaced with a data reduction system (AI450, Dionex).
3.2.10. Separator and guard columns, anion (Model
IonPac®-AS7 and
3.2.11. Syringe prefilters, 0.5-µm pore size (part no. SLSR 025 NS, Millipore Corp., Bedford, MA).
Note: Some syringe prefilters are not cation- or
3.2.12. Scintillation vials, glass, 20-mL.
3.2.13. Equipment for eluent degassing (vacuum pump, ultrasonic bath).
3.2.14 Optional: Centrifuge for spinning down precipitate in samples.
3.3. Reagents - All chemicals should be at least reagent grade. Consult latest material safety data sheets (MSDS) for cautions and proper handling.
3.3.1. Principal reagents:
Sodium carbonate
(Na2CO3), 99%
Sodium bicarbonate (NaHCO3), 99%
Potassium dichromate
(K2Cr2O7),
99.9% or Potassium chromate
(K2CrO4), 99.9%
Magnesium sulfate, anhydrous (MgSO4),
99%
Ammonium sulfate
[(NH4)2SO4
], 99+%
Ammonium hydroxide
(NH4OH), 29%
1,5-Diphenylcarbazide
(DPC) -
C6H5NHNHCONHNHC6H5,
99%
Methanol (CH3OH), HPLC grade
Sulfuric acid
(H2SO4),
concentrated (98%)
Nitric acid (HNO3),
concentrated (69 - 71%)
Deionized water (DI
H2O)
The initial studies were performed using magnesium chloride as the source of magnesium, but this formed a very fine precipitate of magnesium hydroxide. The source of magnesium was switched to magnesium sulfate, because the magnesium sulfate formed a larger sized precipitate which was easier to separate.
3.3.2. Nitric acid, 10% (v/v):
Carefully add 100 mL of concentrated
HNO3 to about
3.3.3. Buffer/extraction (BE) solution (10% Na2CO3 + 2% NaHCO3):
Dissolve 100 g
Na2CO3 and 20 g
NaHCO3 in about 500 mL DI
H2O contained in a
3.3.4. Spray-paint extraction (SPE) solution (5% NaOH + 7.5% Na2CO3):
Dissolve 50 g NaOH and 75 g
Na2CO3 in about
500 mL DI H2O contained in a
3.3.5. Magnesium sulfate solution [~10 mg/mL as Mg(II)]:
Dissolve 9.90 g of anhydrous MgSO4 in 100-mL volumetric flask containing 50 mL DI H2O. Mix well and dilute to the mark with DI H2O. Prepare monthly.
3.3.6. Phosphate buffer (0.5 M KH2PO4/0.5 M K2HPO4· 3H2O):
Dissolve 6.80 g of
KH2PO4 and 11.41
g of
K2HPO4·
3H2O in
3.3.7. Phosphate buffer/Mg(II) (PBM) solution:
Pipette 25 mL of the magnesium sulfate solution (Section 3.3.5.)
into a
3.3.8. Dilute Buffer Extraction/Phosphate buffer/Mg(II) or DBE/PBM solution [for working standard preparation (Section 3.4.)]:
Pipette 50 mL of the BE solution (Section 3.3.3.) into a 100-mL volumetric flask containing 15 mL of PBM solution (Section 3.3.7.). Mix well and dilute to the mark with DI H2O. Magnesium hydroxide will form and precipitate out of solution. Allow the precipitation to settle for at least 60 min., or place in a centrifuge at 3,200 rpm for 10 min. Transfer the "clear" solution to a beaker. Prepare this solution just before working standard preparation.
3.3.9. Eluent [250 mM (NH4)2SO4 + 100 mM NH4OH]:
Dissolve 33 g of (NH4)2SO4 in about 500 mL of DI H2O. Add 6.5 mL of 29% NH4OH. Mix well and dilute with DI H2O to 1.0 L in a volumetric flask. Sonicate this solution and degas under vacuum for 5 min. Transfer the solution to the eluent container.
3.3.10. Postcolumn derivatization reagent (2.0 mM DPC + 10% CH3OH + 1N H2SO4):
1) First dissolve 0.5 g of DPC in 100 mL of HPLC grade
CH3OH. 2) Add 28 mL of 98%
H2SO4 to about
500 mL of DI H2O (CAUTION !!! Make
additions very, very slowly, with mixing, and allow to cool.)
3) Mix solutions (1) and (2) carefully and dilute, with
stirring, in an
3.3.11. Cr(VI) stock standard (100 µg/mL):
Dissolve and dilute 0.2828 g of K2Cr2O7 or 0.3735 g of K2CrO4 to 1.0 L with DI H2O. Prepare this solution every three months.
3.3.12. Cr(VI) standards (10.0 and 1.0 µg/mL):
To prepare 10.0 and 1.0 µg/mL Cr(VI) standards: 1)
Pipette
Note: The laboratory should have an effective, independent quality control (QC) program in place and QC samples of the analyte should be routinely analyzed along with field samples. Depending on the capabilities of the program, QC samples can either be generated using the collection media and chromate compounds under controlled conditions, or media can be spiked with the analyte (such as K2CrO4 or K2Cr2O7). If QC samples cannot be routinely prepared and analyzed, two different standard stock solutions should always be prepared and these solutions should routinely be compared to each other. Always prepare the stocks from two different sources or, as last resort, from different lots.
3.4. Working Standard Preparation - Prepare weekly.
Prepare Cr(VI) working standards in "clear" DBE/PBM solution. A
suggested scheme for preparing a series of working standards using
| Working
Std (ng/mL) |
Std
Solution (µg/mL) |
Aliquot (µL) |
DBE/PBM Added (mL) | |||
| 1.0 5.0 10.0 20.0 50.0 100.0 200.0 500.0 1000.0 |
1.0 1.0 1.0 10.0 10.0 10.0 100.0 100.0 100.0 |
10.0 50.0 100.0 20.0 20.0 100.0 20.0 50.0 100.0 |
9.99 9.95 9.90 9.98 9.95 9.90 9.98 9.95 9.90 | |||
Serial dilutions with volumetric pipets and volumetric flasks may be used instead of a micropipette.
3.5. Sample Preparation
3.5.1. Wash all glassware in hot water with detergent and rinse with tap water, 10% HNO3, and DI H2O (in that order). Caution: Under no circumstances should chromic acid cleaning solutions be used.
3.5.2. Adjust the hot plate to a temperature below the boiling point of the BE solution. A plate surface temperature near 135°C is adequate for extraction. If the hotplate cannot be adjusted to 135°C, use a hot water bath.
3.5.3. If bulk samples are submitted, weigh out a representative aliquot of each bulk on separate PVC blank filters. The bulk and PVC filters are placed in a beaker or flask. To prevent potential future contamination, a beaker or flask of larger size than the air samples should be used.
3.5.4. Carefully remove each PVC filter from their cassettes or
balance, place them
Note: Always add PBM solution before adding the extraction solution. The freshly precipitated magnesium hydroxide [10 mg of Mg(II)] formed suppresses the oxidation of dissolved Cr(III) to Cr(VI) (see Section 4.4. for details).
Swirl the beaker slowly until the white precipitate occurs. Cover
the beaker with a watch glass and very slowly heat the solution on
the hot plate (surface temperature near 135 °C), with occasional
swirling for 60 to 90 min. Allow extra extraction time for heavily
loaded samples taken from
3.5.5. Allow the solutions to cool to room temperature.
Quantitatively transfer each solution to a
3.5.6. If the solution is cloudy and/or other metal analyses are desired, filter the solution through a syringe prefilter. Alternately, cloudy samples may be centrifuged at 3,200 rpm for 10 min. to precipitate the MgOH. Avoid transferring any of the precipitate to the autosampler vials, as it will clog the IC autosampler, tubing, and/or column frits.
3.5.7. FOR SAMPLES TAKEN FROM SPRAY-PAINTING OPERATIONS ONLY, PERFORM AN ADDITIONAL EXTRACTION OF EACH FILTER CONTAINING THE PAINT RESIDUE ACCORDING TO THE FOLLOWING PROCEDURE:
| Note: | Evidence indicates stronger base extractions are capable
of recovering Cr(VI) in specific paint matrices (5.4.). Due to
the resistant properties of some industrial paints, an
additional extraction is used for samples collected during
|
3.6. Analysis
3.6.1. Pipette a 0.5- to 0.6-mL "clear" portion of each standard
or sample solution into separate automatic sampler vials (Note: Be
careful not to transfer any of the
3.6.2. Load the automatic sampler with labeled samples, standards, and blanks.
3.6.3. Set up the ion chromatograph in accordance with the
Standard Operating Procedure (SOP) (5.19.). A diagram of the system
flow path (adapted from Reference 5.12.) is shown in Figure 1.
Typical operating conditions for a Dionex 4000i, 4500i, or DX500
with a
Note: An SOP is a written procedure for a specific instrument. It is suggested that SOPs be prepared for each type of instrument used in a lab to enhance safe and effective operation.
| Ion Chromatograph | |
| Eluent: | 250 mM (NH4)2SO4 /100 mM NH4OH |
| Postcolumn reagent: | 2 mM DPC/10% CH3OH/1 N H2SO4 |
| Column temperature: | ambient |
| Anion precolumn: | IonPac NG1 |
| Anion separator column: | IonPac AS7 |
| Output range: | 0.5 absorbance unit full scale (AUFS) |
| Detection wavelength: | 540 nm |
| Sample injection loop: | 100 µL (a 150 µL loop was used on the DX-500) |
| Pump | |
| Pump pressure: | ~950 psi |
| Eluent flow rate: | 0.7 mL/min |
| Postcolumn reagent flow rate: | ~0.34 mL/min |
| Chromatogram: | |
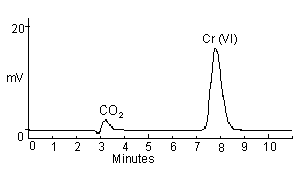 A chromatogram of 100 ng/mL Cr(VI). The CO2 peak is from the reaction of the bicarbonate and carbonate ions with the sulfuric acid in the post column derivatization mixture. The size of the CO2 peak can be changed or eliminated by the amount of back pressure on the waste line coming from the detector. | |
| Run time: | 11 min |
| Peak retention time: | ~8 min for Cr(VI) (Please note that peak retention times are highly dependent on and individualized to the instrument in use and the age of the column.) |
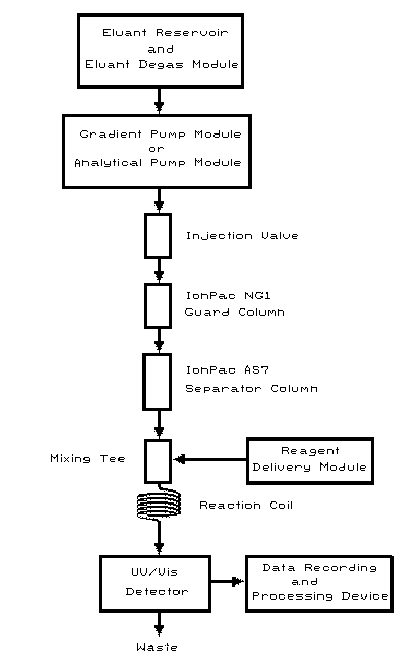
Figure 1. A diagram
of the system flow path.
3.6.4. Follow the SOP for further instructions regarding analysis (5.19.).
3.6.5. If any sample has a Cr(VI) concentration larger than the highest standard, dilute the sample by taking an appropriate aliquot and add an appropriate amount of DBE/PBM solution to bring the sample concentration within the range of the standards. A dilution factor (DF) as calculated from the aliquot volume and diluent volume is used in final calculations (e.g., if a 2 mL aliquot is taken and 8 mL of DBE/PBM is added, then a DF of 5 is used.)
3.7. Calculations
3.7.1. After the analysis is completed, retrieve the peak areas or heights. Obtain hard copies of chromatograms from a printer.
3.7.2. Prepare a concentration-response curve by plotting the peak areas or peak heights versus the concentration of the Cr(VI) standards in ng/mL. Peak areas are preferred. Typical instrumental response for working standards from 10 to 1000 ng/mL range using a Dionex Model DX500 equipped with an AD20 Absorbance Detector and GP40 Gradient Pump as follows:
| Level | Concentration ng/mL |
Peak Height (× 104) |
Peak Area (× 106) |
| 1 | 0.500 | 0.005 | 0.015 |
| 2 | 1.000 | 0.010 | 0.031 |
| 3 | 5.000 | 0.051 | 0.152 |
| 4 | 10.00 | 0.095 | 0.279 |
| 5 | 20.00 | 0.190 | 0.545 |
| 6 | 50.00 | 0.491 | 1.422 |
| 7 | 100.0 | 0.980 | 2.803 |
| 8 | 200.0 | 1.858 | 5.245 |
| 9 | 500.0 | 4.522 | 12.363 |
| 10 | 1,000 | 9.628 | 24.736 |

Figure 2. A plot of
the standard curve of the above standards.
3.7.3. Perform a blank correction for each PVC filter result. Subtract the ng/mL Cr(VI) blank value (if any) from each sample reading if blank and sample solution volumes are the same. If a different solution volume is used, subtract the total ng blank value from each total ng sample value.
Ab = [ng/mL Cr(VI)]b × (Sol Vol)b
As = [ng/mL Cr(VI)]s × (Sol Vol)s
A = [As - Ab] × DF
Then calculate the air concentration of Cr(VI) (in µg/m3) for each air sample:
| µg/m3 Cr(VI) = |
|
| Where: | ||
| Ab | = | Total ng Cr(VI) in blank |
| As | = | Total ng Cr(VI) in sample |
| A | = | ng Cr(VI) after blank correction |
| [ng/mL Cr(VI)]b | = | Amount found (from calibration curve) in blank |
| [ng/mL Cr(VI)]s | = | Amount found (from calibration curve) in sample |
| (Sol Vol)b | = | Blank solution volume (mL) from Section 3.5.5. (normally 10 mL*) |
| (Sol Vol)s | = | Sample solution volume (mL) from Section 3.5.5. (normally 10 mL*) |
| AV | = | Air volume (L) |
| DF | = | Dilution factor (if any, see Section 3.6.5.) |
| = | (mL Diluent + mL Aliquot)/mL Aliquot | |
| *The solution volume for each SPE sample is normally 25 mL. | ||
3.7.4. For bulk samples, calculate the total composition (in %) of Cr(VI) in each sample using:
| % (w/w) Cr(VI) = |
|
| Where: | ||
| A | = | µg Cr(VI) after blank correction |
| D | = | Dilution factor (if any) |
| SW | = | Aliquot (in mg) of bulk taken in Section 3.5.3. |
3.8. Reporting Results
3.8.1. For spray-paint samples, add results obtained from the SPE residue extraction, if any, to the initial extraction result.
3.8.2. Report air sample results to the industrial hygienist as µg/m3 Cr(VI).
3.8.3. Report wipe sample results to the industrial hygienist as total micrograms or milligrams.
3.8.4. Report bulk sample results to the industrial hygienist as approximate per cent Cr(VI).
4. Backup Data
This method has been validated using a full shift sample of 480-min
taken at a flow rate of 2 L/min for a
The validation consisted of the following experiments and discussion:
- An analysis of 18 spiked (soluble chromate) samples - This set
consists of 6 samples each at 0.25 ×, 0.5 ×, and 1 × the proposed OSHA
TWA-PEL assuming960-L air volumes, to determine bias, precision, and overall error (OE) (Note: One sample at 1× PEL was lost during analysis). - An analysis of 18 spiked (insoluble chromate) samples - This set
consists of 6 samples each at 0.25 ×, 0.5 ×, and 1 × the proposed OSHA
TWA-PEL assuming960-L air volumes, to determine bias, precision, and OE. - An evaluation of storage stability at room temperatures (20 to 25 °C) for 24 spiked samples.
- A determination of the qualitative and quantitative detection limits for Cr(VI).
- An interference study using varied amounts of reducing substances and addition of Mg (II) to prevent oxidation of Cr(III) to Cr(VI).
- A comparison of BE dilutions using concentration ratios (v:v) of 1:10, 1:8, 1:5 and 1:2.
- An analysis of 3 sets of Cr(VI) quality control (QC) samples.
- An evaluation of a strong extraction solution for
spray-paint samples. - An analysis of Cr(VI) field samples using both DPP and
IC/
UV-vis methods. - A summary.
An aerosol generation system to determine sampler efficiency was
unavailable; however, this method (OSHA
OEi% = ±(|biasi| + 2CVi) × 100% (at the 95% confidence level)
Where i is the respective sample pool being examined.
4.1. Spiked Sample Analysis
Samples were prepared by adding known amounts of K2Cr2O7 and PbCrO4 stock solutions to PVC filters (also see Section 4.2. for preparation) to determine bias, precision, and OE for the analytical portion of the method. Samples were prepared with and without the addition of phosphate buffer/Mg(II) to evaluate any difference in recoveries. The lower concentration, 0.25 × and 0.5 × TWA PEL were used for this comparison.
4.1.1. Procedure: The PVC filters were spiked using
a 25-µL syringe (Hamilton
Microliter®/Gastight® Syringe, Hamilton Co.,
Reno, NV). Spikes (both
K2Cr2O7
and PbCrO4) were 0.11, 0.20, and 0.40
µg as Cr(VI). These levels correspond approximately to 0.25,
0.5, and 1 × the proposed OSHA TWA PEL for a
4.1.2. Results: Recoveries are presented in Tables 1a, 1b, and 1c. As shown, including addition of phosphate buffer/Mg(II) in Table 1c, the mean recovery for all levels tested is very close to 1.0 for both soluble and insoluble chromate compounds. No DE corrections are necessary for Cr(VI) collection using PVC filters.
| Level | N | Mean Recovery |
SD | CV | OET ±% |
| 0.25 × PEL | 6 | 1.047 | 0.061 | 0.058 | 16.3 |
| 0.5 × PEL | 6 | 1.002 | 0.061 | 0.061 | 12.4 |
| 1 × PEL | 5* | 0.966 | 0.035 | 0.037 | 10.9 |
| All Levels | 17 | 1.007 | 0.054 | 11.5 |
*One sample was lost in analysis.
| Level | N | Mean Recovery |
SD | CV | OET ±% |
| 0.25 × PEL | 6 | 1.019 | 0.079 | 0.078 | 17.5 |
| 0.5 × PEL | 6 | 0.970 | 0.021 | 0.021 | 7.2 |
| 1 × PEL | 6 | 0.969 | 0.074 | 0.076 | 18.3 |
| All Levels | 18 | 0.986 | 0.064** | 14.2 |
** = CV1 (pooled)
| Level | N | Mean Recovery |
SD | CV | OET ±% |
| 0.25 × PEL | 6 | 1.000 | 0.112 | 0.112 | 22.4 |
| 0.5 × PEL | 7 | 0.985 | 0.007 | 0.008 | 3.1 |
| Where | ||
| N = Number of Samples; | SD = Standard Derivation | |
| CV = Coef. of Variation; | OET = Overall Error (Total) |
4.2. Storage Stability
Procedure: Twenty-four samples were spiked to
evaluate stability prior to sample analysis. A
PbCrO4 stock solution was used to spike
samples near 0.5 × the proposed OSHA TWA PEL [as Cr(VI)] for a
Another storage experiment was also conducted using prepared
extraction solutions with DBE and phosphate buffer/Mg(II). This
experiment was performed separately to evaluate storage after the
samples were prepared. Six samples were spiked using the soluble
K2Cr2O7
stock solution at 0.25 × the proposed OSHA TWA PEL [as Cr(VI)] for a
Results: As shown in Tables 2a and 2b, the results of both tests conducted at room temperature show the mean recovery from filter and extracted samples analyzed after 30 days was within ±5% of the recovery value at day 0.
| Day | N | Mean, µg | SD | CV | Recovery (%) |
| 0 | 6 | 0.197 | 0.004 | 0.021 | 97.0 |
| 5 | 6 | 0.190 | 0.005 | 0.026 | 93.6 |
| 15 | 6 | 0.200 | 0.018 | 0.088 | 98.7 |
| 30 | 6 | 0.190 | 0.008 | 0.040 | 93.7 |
| Day | N | Mean, µg | SD | CV | Recovery (%) |
| 0 | 6 | 0.120 | 0.013 | 0.11 | 100 |
| 30 | 6 | 0.126 | 0.010 | 0.08 | 105 |
4.3. Qualitative and Quantitative Detection Limit Study
A modification of the National Institute for Occupational Safety and Health (NIOSH) detection limit calculation procedure (5.24., 5.25.) was used to calculate detection limits.
Procedure: Ten different concentrations were used by
spiking six separate PBM/DBE solutions (Section 3.3.8.) with aliquots
of aqueous standards prepared from
K2Cr2O7
(Section 3.3.11.). All samples were analyzed using a 100-µL
sample injection loop and a
Results: The spiked sample results are shown in Table
3 for qualitative and quantitative detection limits, respectively. The
qualitative detection limit was 1 ng [as Cr(VI)] when using a
|
| ||||||||||
| ----------- Cr(VI) Level (as ng/mL)------ | ||||||||||
| Sample number |
0.1 PA |
0.2 PA |
0.3 PA |
0.4 PA |
0.5 PA |
0.6 PA |
0.7 PA |
0.8 PA |
0.9 PA |
1.0 PA |
| 1 | 1644 | 4786 | 7292 | 11136 | 15252 | 17612 | 19970 | 23583 | 29116 | 31324 |
| 2 | 1726 | 4911 | 7264 | 11143 | 15772 | 17188 | 19978 | 23190 | 29956 | 31414 |
| 3 | 1774 | 4933 | 7319 | 11575 | 15510 | 17412 | 19725 | 23444 | 29348 | 31402 |
| 4 | 1742 | 4999 | 7486 | 11576 | 14859 | 16850 | 21384 | 23667 | 29237 | 31697 |
| 5 | 1436 | 4862 | 7017 | 11553 | 14530 | 17528 | 21658 | 23519 | 29289 | 30908 |
| 6 | 1748 | 4902 | 7039 | 11675 | 15404 | 16978 | 21638 | 23680 | 30207 | 31968 |
|
| ||||||||||
| PA = Integrated Peak Area The blank integrated peak areas and their standard deviations (std dev) were all equal to zero. | ||||||||||
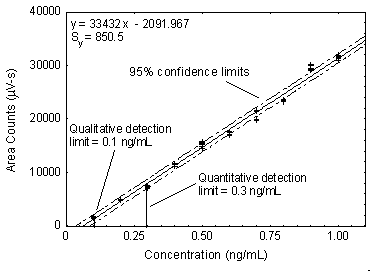
Figure 3. A Plot of
the standards to determine the detection limit.
The response of the low-level calibration samples were plotted to
obtain the linear regression equation (Y = mX + b), and the predicted
responses ( i) at each X.
i) at each X.
| Using the equations: | Sy = [S(i -
Yi)2/(N
- 2)]½ |
| Q1 = (3Sy)/m | |
| Q2 = 3.33 Q1 |
Where:
| Yi | = the measured response |
| m | = analytical sensitivity or slope as calculated by linear regression |
| Sy | = the standard error of the regression |
| N | = the number of data points |
| Q1 | = qualitative detection limit |
| Q2 | = quantitative detection limit |
| Therefore, | Q1 | = | (3Sy)/m |
| = | 0.1 ng/mL as Cr(VI) | ||
| |
1.0 ng as Cr(VI) (10-mL sample volume) | ||
| |
1.0 × 10-3 µg/m3 as Cr(VI) (960-L air volume) | ||
| Q2 | = | 3.33 Q1 | |
| |
3.0 × 10-3 µg/m3 as Cr(VI) (960-L air volume) | ||
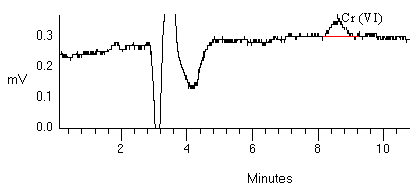
Figure 4. A
chromatogram of the quantitative detection limit of 0.3 ng/mL Cr(VI).
It is interesting to note that the addition of phosphate buffer/Mg(II) to the solutions significantly increased detection limits. The qualitative and quantitative limits without addition of the Cr(III) conversion suppressor were approximately six times less than the limits stated above. In standards above 50 ng/mL this difference was not noted. In standards less than 50 ng/mL the difference between standards prepared with only BE and those with the addition of phosphate buffer/Mg(II) increases as the concentration of the standards decreases, such that the lower end of the calibration curve becomes quadratic.
4.4. Interference Study
Six experiments to test potential interferences from various amounts of Cr(III), Fe(II), Fe(III), V(V), Mo(VI), Cu(I), and Mn(II) were conducted. These substances may coexist with Cr(VI) compounds in some workplace atmospheres and may also interfere with the analysis of Cr(VI) (5.3.). The following chemicals were used for preparing the solution spikes for this study:
Potassium dichromate, K2Cr2O7, for Cr(VI); Chromium nitrate, Cr(NO3)3 9H2O, for Cr(III); Ferrous sulfate, FeSO4, for Fe(II); Ferric nitrate, Fe(NO3)3, for Fe(III); Vanadium pentoxide, V2O5, for V(V); Molybdenum trioxide, MoO3, for Mo(VI); Cuprous chloride, Cu2Cl2, for Cu(I); Manganous chloride, MnCl2 4H2O, for Mn(II); and Magnesium chloride, MgCl2, or Magnesium sulfate, MgSO4, for Mg(II).
All Cr(III) solutions were used to test how much, if any, Cr(III) converts to Cr(VI) on PVC filters or in solution. Mixtures using Mg(II) were used to determine its ability to suppress potential interferences. Early experiments were conducted using magnesium chloride to provide the magnesium needed to form the magnesium hydroxide precipitate with any Cr(III) present. Magnesium sulfate was also used in a comparison between the two salts in an extraction study. Both the chloride and the sulfate of magnesium gave comparable results. Magnesium sulfate is recommended in this method because of the better, larger precipitate formation. A significant difference between the two salts was not noted in terms of recovery, peak characteristics, or retention times. A difference was noted in that the magnesium chloride gave a precipitate that was more difficult to decant. The six experiments are detailed in Sections 4.4.1. through 4.4.6. below.
4.4.1. Differing amounts of Cr(VI) and each of the interfering substances were mixed in the same volumetric flasks and then spiked onto individual PVC filters. The concentrations of the spikes varied from 0 to 10 times the Cr(VI) concentration.
Procedure: Fifteen different potential interference mixture combinations and six samples of each combination were prepared, extracted with BE, and analyzed after 1:1 dilution. A large amount (887.6 and 872 ng/mL) of Cr(VI) was used for the spikes in this Experiment (and also Experiment 3) so that any significant effect would be analytically obvious.
Results: The recoveries for Cr(VI) with varied amounts of reducing substances are shown in Table 4a.
| No. | Mixture Composition |
Ratio | N | Mean, ng/mL |
SD | CV | Recovery, % As Cr(VI) |
| 1 | Cr(VI) only | 1:0 | 6 | 887.6 | 26.0 | 0.029 | 100 |
| 2 | Cr(VI):Cr(III) | 1:10 | 6 | 911.5 | 23.5 | 0.026 | 103 |
| 3 | Cr(VI):Fe(II) | 1:10 | 6 | 258.9 | 8.79 | 0.034 | 29.2 |
| 4 | Cr(VI):Fe(III) | 1:10 | 6 | 918.5 | 19.5 | 0.021 | 103 |
| 5 | Cr(VI):V(V) | 1:10 | 5 | 915.8 | 29.7 | 0.032 | 103 |
| 6 | Cr(VI):Mo(VI) | 1:10 | 6 | 874.5 | 16.6 | 0.019 | 98.5 |
| 7 | Cr(VI):Cu(I) | 1:10 | 6 | 898.0 | 76.4 | 0.085 | 101 |
| 8 | Cr(VI):Mn(II) | 1:10 | 6 | 838.0 | 33.9 | 0.040 | 94.4 |
| 9 | Cr(VI):Fe(II) | 1:1 | 6 | 811.1 | 18.1 | 0.022 | 91.4 |
| 10 | Cr(VI):Fe(II) | 1:5 | 6 | 643.8 | 12.5 | 0.019 | 72.5 |
| 11 | Cr(VI):Cr(III):Fe(II) | 1:1:1 | 6 | 848.5 | 17.5 | 0.021 | 95.6 |
| 12 | Cr(VI):Cr(III):Fe(II) | 1:5:5 | 6 | 566.3 | 15.9 | 0.028 | 63.8 |
| 13 | Cr(VI):Cr(III):Fe(II) | 1:10:10 | 6 | 291.5 | 10.0 | 0.034 | 32.8 |
| 14 | Cr(VI):Cr(III):Fe(II):
Fe(III):V(V):Mo(VI) |
1:1:1: 1:1:1 |
6 | 841.5 | 11.8 | 0.014 | 94.8 |
| 15 | Cr(VI):Cr(III):Fe(II):
Fe(III):V(V):Mo(VI) |
1:10:10: 10:10:10 |
6 | 761.6 | 30.8 | 0.040 | 85.8 |
As shown above, except for the solution containing large amounts
of Fe(II) over Cr(VI), the recovery range is very close to 100%.
When Cr(III) was added to Fe(II) and Cr(VI) the recovery is 91%, as
shown in samples in set no. 9. Cr(III) added to 1:5 Cr(VI):Fe(II)
had a recovery of 64%, as shown in samples in set no. 12. Cr(III)
added to 1:10 Cr(VI):Fe(II) had recovery of 33%, as shown in samples
in set no. 13. These losses occurred in a slightly acidic
environment [both analytes were prepared in DI
H2O (pH![]() 5.5) and contained
in the same volumetric flask].
5.5) and contained
in the same volumetric flask].
4.4.2. Once the Fe(II) interference was identified in Experiment 1, a smaller amount of Cr(VI) and Fe(II) were used for Experiment 2. An additional test was performed to determine conversion of Cr(III) to Cr(VI).
Procedure: Cr(VI) was spiked onto PVC filters first, dried, and then differing amounts of Fe(II) or Cr(III) were spiked on the Cr(VI) spot, dried, and then extracted with BE, and analyzed after 1:1 dilution.
Results: Table 4b shows the recoveries for Cr(VI) are close to 70% for 1:1, 1:5, and 1:10 Cr(VI) : Fe(II). This approximately 30% loss apparently occurred while both spikes were residing on the filter. A very small amount of Cr(III) converting to Cr(VI) is noted in Table 4b (0.71 ng/mL).
| No. | Mixture Composition |
Ratio or Amount | N | Mean, ng/mL |
SD | CV | Recovery, % As Cr(VI) |
| 1 | Cr(VI) only | 101.5 ng/mL Cr(VI) | 6 | 101.5 | 3.72 | 0.037 | 100 |
| 2 | Cr(III) only | 1.0 µg/mL Cr(III) | 6 | 0.71* | 0.36 | 0.50 | <1* |
| 3 | Cr(VI):Fe(II) | 1:1 | 5 | 72.0 | 4.41 | 0.061 | 70.9 |
| 4 | Cr(VI):Fe(II) | 1:5 | 5 | 69.2 | 6.66 | 0.096 | 68.2 |
| 5 | Cr(VI):Fe(II) | 1:10 | 6 | 69.0 | 5.24 | 0.076 | 68.0 |
*Cr(III) converted to Cr(VI)
4.4.3. The SPE solution, which contained 5% NaOH and 7.5% Na2CO3, was used as an extraction solution in Experiment 3 to evaluate the ease of converting Cr(III) to Cr(VI) in a stronger base. The experiment was also conducted to test whether or not magnesium (Mg) can prevent conversion of Cr(III) to Cr(VI) in SPE solutions. This conversion was noted in the literature (5.6.) when using a NaOH/Na2CO3 extraction similar to SPE, but was not noted in earlier work using BE solutions (5.8.), primarily because of the significantly higher PEL and spiking concentrations used.
Procedure: Cr(VI) was spiked onto PVC filters first, dried, and then Cr(III) was spiked on the Cr(VI) spot, dried, and then extracted with SPE, and analyzed after 1:1 dilution.
Results: Table 4c shows adding 1 mg of Mg(II) can prevent Cr(III) converting to Cr(VI). This was the same conclusion presented in reference 5.6.
| No. | Mixture Composition |
Ratio or Amount | N | Mean, ng/mL |
SD | CV | Cr(III) Converted to Cr(VI), % |
| 1 | Cr(VI) only | 872 ng/mL Cr(VI) | 4 | 872 | 16 | 0.018 | - |
| 2 | Cr(III) only | 10 µg/mL Cr(III) | 4 | 18* | 1.3 | 0.069 | <0.2 |
| 3 | Cr(VI):Cr(III) | 1:10 | 4 | 880 | 12 | 0.013 | <0.1 |
| 4 | Cr(III) + 1 mg Mg(II) | 10 µg/mL Cr(III) | 4 | ND | - | - | - |
| 5 | Cr(VI):Cr(III) + 1 mg Mg(II) | 1:10 | 4 | 1055 | 10 | 0.012 | <0.03 |
*Cr(III) converted to Cr(VI)
Note: ND = 0.251
ng/mL as Cr(VI)
4.4.4. Experiment 4 was conducted to further test the effectiveness of Mg(II) with large proportions of Cr(III) to Cr(VI) in both BE and SPE solutions. Because Cr(VI) is significantly more toxic than Cr(III) [Note: The TWA PELs for Cr(VI) and Cr(III) are 0.50 µg/m3 (proposed) and 1 mg/m3, respectively], the concentration ratio of Cr(VI) and Cr(III) in Experiment 4 was: Cr(VI) : Cr(III) = 250 ng: 5 mg = 1: 20,000.
Procedure: Experiment 4 included 10 tests. The
first 5 tests were conducted using BE solution and the last 5 tests
were conducted using SPE solution. Each sample was spiked with 250
ng of Cr(VI) or 5 mg of Cr(III) while contained in a
A: 250 ng of Cr(VI) (control samples);
B: 5 mg
of Cr(III) [check for conversion to Cr(VI) during
extraction];
C: 250 ng of Cr(VI) + 5 mg of
Cr(III);
D: C + 10 mg Mg(II);
E: C + 20 mg Mg
(II);
A': 250 ng of Cr(VI) (control samples);
B':
500 mg of Cr(III) [check for conversion to Cr(VI) during
extraction];
C': 250 ng of Cr(VI) + 5 mg of
Cr(III);
D': C' + 10 mg Mg(II);
E': C' + 20 mg
Mg(II);
Results: Table 4d data suggests that the oxidation of Cr(III) occurred during the alkaline extraction process. When alkalinity was increased by using 5% NaOH, more Cr(III) was oxidized to Cr(VI) (as shown in SPE, Samples A' to E'). Although the conversion is small as percentage of Cr(III), it is very significant in terms of the proposed PEL. A previous work conducted by the author (5.8.) did not note the conversion in BE solutions; however, the larger detection limit and lack of significance (the PEL of 0.05 mg/m3 was used in the past work) were contributing factors. The net conversion of Cr(III) to Cr(IV) is considered extremely minor when comparing amounts to the PEL of 0.05 mg/m3. In the presence of freshly precipitated magnesium hydroxide (10 or 20 mg of Mg) the oxidation of dissolved Cr(III) was suppressed to insignificantly low levels. As shown in Table 4d, the approach with Mg(II) is also applicable in the more strongly basic solution of SPE (5% NaOH/7.5% Na2CO3). It should be noted that the SPE extraction is performed after the BE extraction, and little, if any, soluble Cr(III) should still be present. It is important to note, for maximum effectiveness, the magnesium salt/phosphate buffer solution is added to the sample before BE or SPE solutions.
| Set # | N | ng Cr(VI) found theoretical = 250 ng |
SD | CV | Cr(III) converted to Cr(VI)(%) |
| A | 6 | 249.57 | 3.98 | 0.016 | - |
| B | 6 | 128.03 | 7.93 | 0.062 | 0.00256 |
| C | 6 | 373.19* | 7.74 | 0.021 | 0.00246 |
| D | 6 | 250.07* | 5.27 | 0.021 | - |
| E | 6 | 237.82* | 2.97 | 0.013 | - |
| A' | 6 | 253.06 | 3.60 | 0.014 | - |
| B' | 6 | 226.45 | 8.23 | 0.036 | 0.0045 |
| C' | 6 | 484.79* | 13.07 | 0.027 | 0.0047 |
| D' | 6 | 281.43* | 5.12 | 0.018 | 0.00063 |
| E' | 6 | 268.18* | 6.17 | 0.023 | 0.00036 |
*Cr(III) converted to Cr(VI) plus 250 ng Cr(VI) spike
4.4.5. Experiment 5 was conducted to repeat certain aspects of Experiment 4 and to determine the amount of Mg(II) needed to prevent Cr(III) conversion to Cr(VI) during the extraction process.
Procedure: Experiment 5 repeated the design of Experiment 4, except that Cr(VI) : Cr(III) = 500 ng : 5 mg = 1:10,000. The following sets used in this experiment are (Mg(II) is as MgSO4):
F: 500 ng of Cr(VI) + 5 mg of Cr(III) + 5 mg Mg(II) with
BE;
G: 500 ng of Cr(VI) + 5 mg of Cr(III) + 10 mg Mg(II)
with BE;
H: 500 ng of Cr(VI) + 5 mg of Cr(III) + 15 mg
Mg(II) with BE;
G': 500 ng of Cr(VI) + 5 mg of Cr(III) +
10 mg Mg(II) with SPE;
Results: Table 4e shows that, in BE solution, the addition of 5, 10, or 15 mg of Mg(II) to a mixture of Cr(III) and Cr(IV) gave comparable results. The slight decrease in recovery as Mg(II) increased appears more so as noise resulting from analyzing a very small amount (500 ng) of Cr(VI). It was noted that the addition of Mg(II) produces a significant precipitate of magnesium hydroxide in the extraction solution and that the more added, the larger the precipitate. This precipitate must be carefully handled when transferring solutions for analysis to prevent injection into the ion chromatograph.
| Set # | N | Mean ng as
Cr(VI) Theory=500ng |
SD | CV | Cr(III) converted to Cr(VI), % |
| F | 6 | 507.55* | 2.88 | 0.0057 | <0.01 |
| G | 6 | 496.59* | 3.67 | 0.0074 | - |
| H | 6 | 497.35* | 5.82 | 0.0096 | - |
| G' | 6 | 508.48* | 4.86 | 0.0096 | <0.01 |
*Cr(III) converted to Cr(VI) plus 500 ng Cr(VI) spike.
4.4.6. Experiment 6 was performed to test whether or not adding Mg(II) or phosphate buffer (0.5 M KH2PO4/0.5 M K2HPO4)/Mg(II) can also prevent the negative Fe(II) interference on Cr(VI) analysis. The phosphate buffer is thought to aid in complexing the Cr(III) (5.5.).
Procedure: Experiment 6 included 2 tests. The first test was conducted using only Mg(II) spiking on Fe(II); the second test was performed using the mixture of phosphate buffer/Mg(II) on the Fe(II). A known amount of Cr(VI) was spiked on one side of each PVC filter and the Fe(II) spiked on the other side of each filter. The filters were allowed to dry overnight and then Mg(II) or the mixture of phosphate buffer/Mg(II) was added prior to extraction with BE solution. The following sets were used for this experiment:
| I: | 100 ng/mL of Cr(VI) + 1.0 g/mL of Fe(II) + 10 mg Mg(II)(as MgCl2) |
| J: | 100 ng/mL of Cr(VI) + 1.0 g/mL of Fe(II) + 10 mg Mg(II)(as MgCl2 mixed with phosphate buffer). |
| K: | 100 ng/mL of Cr(VI) + 1.0 g/mL of Fe(II) + 10 mg Mg(II) (as MgSO4 mixed with phosphate buffer). |
Results: Table 4f shows a significant increase in recovery of Cr(VI) as compared to Experiment 2 is noted when adding Mg(II) or phosphate buffer/Mg(II) mixture.
| Set # | Mixture Composition |
Ratio | N | Mean, ng/mL | SD | CV | Recovery, % As Cr(VI) |
| I | Cr(VI):Fe(II) | 1:10 | 6 | 92.7 | 4.29 | 0.046 | 92.7 |
| J | Cr(VI):Fe(II) | 1:10 | 6 | 96.6 | 3.41 | 0.035 | 96.6 |
| K | Cr(VI):Fe(II) | 1:10 | 6 | 95.8 | 1.59 | 0.026 | 95.8 |
4.5. Comparison of Different DBE Solutions
Due to the strongly basic nature of the BE solution, a dilution with DI H2O needs to be performed prior to analysis. To determine the most effective dilution, the following experiment was performed.
Procedure: In order to compare the performance of this method and to potentially increase the analytical sensitivity, different DBE solutions were used for testing. Four DBE solutions were prepared from the original BE solution: 1) 1 to 10 dilution of original BE solution; 2) 1 to 8 dilution; 3) 1 to 5 dilution; and 4) 1 to 1 dilution. A spike of 80 ng/mL Cr(VI) was added to each dilution.
Results: Table 5 shows results of the comparison study. As shown, there were no significant differences among the recoveries, however; certain characteristics of the chromatogram changed as the concentration of BE changed.
| Dilution Factor 1 to x |
N | Mean Cr(VI) µg |
SD | CV | Ratio µg(1 to x)/µg(1 to 10) |
| 1 to 10 | 6 | 77.5 | 3.6 | 0.047 | - |
| 1 to 8 | 6 | 80.6 | 1.7 | 0.021 | 1.04 |
| 1 to 5 | 6 | 76.5 | 2.9 | 0.037 | 0.99 |
| 1 to 1 | 5 | 77.3 | 3.5 | 0.046 | 1.00 |
An additional test was performed to assess the differences in the chromatogram using 100 ng/mL Cr(VI) standard in DI H2O, in a 1:1 dilution, and in BE. As shown in the following figure, a peak appearing just before the Cr(VI) peak becomes larger as the concentration of DBE solution becomes stronger, though the size of this peak also depends on the freshness of the DBE/PBM solution, the age of the standards or samples, and the backpressure of the pumps. Broadening of the Cr(VI) peak also occurs, indicating that matrix matching of the standards and samples is necessary. A dilution of 1:1 was chosen to maintain adequate sensitivity with minimal peak broadening when compared to aqueous standards.
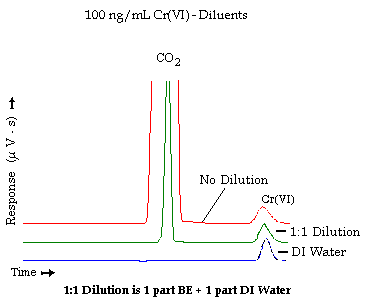
Figure 5. Overlapping chromatograms illustrating the effect of the amount of carbonate/magnesium/phosphate solution on these chromatograms.
4.6. Evaluation of Extraction Solution for Spray-Paint Samples
Procedure: The resistance of
Results: Table 6 shows the results of a comparison of
the effectiveness of these two extraction solutions. As shown, the SPE
solution is superior to the BE solution for extraction of Cr(VI) in
potentially resistant
| Extraction Solution |
N | Mean, µg | SD | CV | Ratio, SPE/BE |
| BE | 5 | 142 | 9.9 | 0.069 | - |
| SPE | 5 | 211 | 33 | 0.16 | 1.49 |
4.7. Comparison of Extraction with MgCl2 and MgSO4
Procedure: PVC filters were spiked with 1 µg Cr(VI) and extracted with a solution of 10 mg/mL Mg(II), in the form of either MgCl2 or MgSO4, in the phosphate buffer, and then BE solution was added.
Results: Table 7 shows that there was little difference in the extraction efficiency between the two different salts of magnesium.
| Type of Mg | N | Mean ng Cr(VI) | SD | CV | Recovery, % |
| MgCl2 | 6 | 1000 | 2.31 | .063 | 100 |
| MgSO4 | 6 | 991 | 1.46 | .042 | 99.1 |
4.8. Analysis of Cr(VI) Quality Control (QC) Samples
Procedure: Three sets of Cr(VI) QC samples were prepared by an independent source by spiking 10 to 20 µg Cr(VI) on the PVC filters. Samples were analyzed using the conditions stated in Section 3 of this method.
Results: Table 8 shows the results of the QC samples, which have amounts typical of those near or over the PEL of 0.05 mg/m3 Cr(VI). Samples with higher concentrations can be analyzed using this method provided higher standards are prepared to bracket the samples, or the appropriate aliquot/dilution is performed.
| Set | N | Mean, F/T* | SD | CV | Recovery, % |
| I | 4 | 0.949 | 0.019 | 0.020 | 94.9 |
| II | 4 | 0.978 | 0.050 | 0.051 | 97.8 |
| III | 4 | 0.940 | 0.049 | 0.053 | 94.0 |
| 0.044** | 95.6 ave. |
* F/T
= Found/Theoretical (Recovery)
** CV (pooled)
4.9. Analysis of Cr(VI) Field Samples
Procedure: In order to compare the new
IC/
Results: Table 9 shows the Cr(VI) results in
mg/m3. The DPP results are in parenthesis
for comparison purposes. As shown, both methods are in good agreement
except for a few very low concentrations in which the DPP method gave
"none detected"results. However, for those
| Sample No. | Air Volume, L | ng/mL, Cr(VI) | µg, Cr(VI) | mg/m3, Cr(VI) |
| 01 | 512.0 | ND | ND | ND (ND) |
| 02 | 632.0 | ND | ND | ND (ND) |
| 03 | 602.0 | ND | ND | ND (ND) |
| 04 (Bl) | 0 | ND | ND | ND (ND) |
| 05 | 42.5 | 62.9 | 6.29 | 0.1480 (0.1838) |
| 06 (Bl) | 0 | ND | ND | ND (ND) |
| 07 | 876.0 | 8.98 | 2.25 | 0.0026 (0.0019) |
| 08 | 588.0 | 6.81 | 1.70 | 0.0029 (0.0017) |
| 09 | 802.0 | 9.82 | 2.46 | 0.0031 (0.0023) |
| 10 | 0 | ND | ND | ND (ND) |
| 11 | 799.2 | 13.3 | 3.33 | 0.0042 (0.0039) |
| 12 | 797.0 | 8.85 | 2.21 | 0.0028 (0.0020) |
| 13 | 869.5 | 13.9 | 3.49 | 0.0040 (0.0041) |
| 14 | 827.5 | 19.1 | 4.79 | 0.0058 (0.0059) |
| 15 | 945.6 | 6.84 | 1.71 | 0.0018 (0.0011) |
| 16 | 930.0 | 4.48 | 1.12 | 0.0013 (ND) |
| 17 | 882.0 | 17.4 | 4.35 | 0.0049 (0.0050) |
| 18 | 884.1 | 7.84 | 1.96 | 0.0022 (0.0016) |
| 19 | 887.3 | 6.07 | 1.52 | 0.0017 (ND) |
| 20 | 276.0 | ND | ND | ND (ND) |
| 21 | 392.0 | 5.37 | 1.34 | 0.0034 (ND) |
| 22 (Bl) | 0 | ND | ND | ND (ND) |
| 23 (Wipe) | 0 | 5.09 | 1.27 µg | 1.27 µg (1.06 µg) |
| 24 | 64.3 | 15.4 | 1.54 | 0.0239 (0.0247) |
| 25 | 52.0 | ND | ND | ND (ND) |
| 26 | 181.7 | ND | ND | ND (ND) |
| 27 (Wipe) | 0 | 6.00 | 1.50 µg | 1.50 µg (0.85 µg) |
| 28 (Bl) | 0 | ND | ND | ND (ND) |
| 29 | 63.0 | 4.72 | 0.47 | 0.0075 (ND) |
| 30 | 74.1 | ND | ND | ND (ND) |
| 31 (Bl) | 0 | ND | ND | ND (ND) |
| 32 | 566.0 | ND | ND | ND (ND) |
| 33 | 658.0 | ND | ND | ND (ND) |
Note: For IC/
4.10. Summary
This analytical method has been shown to be precise and accurate when analyzing soluble and insoluble chromate compounds (as potassium dichromate and lead chromate, respectively) commonly found in the workplace. The validation results indicate the method meets the OSHA criteria for accuracy and precision (5.23.). Performance during storage stability tests is adequate. Detection limits [as Cr(VI)] are very low when samples are taken for 8 h at 2 L/min. No significant interferences were found from various amounts of reducing substances except for samples containing Fe(II). Results indicate that not only does the addition of magnesium sulfate or magnesium chloride prevent the conversion of Cr(III) to Cr(VI), but also can minimize the Fe(II) effect on Cr(VI) analysis.
A 1:1 dilution was used for optimal sensitivity. A peak prior to
the Cr(VI) peak is noted, and slight peak broadening occurs with this
dilution; however, as long as matrix matching of standards and samples
occur, significant problems are not noted. The method demonstrates
good performance in analyzing Cr(VI) QC samples and is not only in
good agreement with the DPP technique (OSHA Method No.
5. References
5.1. National Institute for Occupational Safety and Health:
Method No. P&CAM 169, in NIOSH Manual of Analytical
Methods. 2nd ed., Vol. 1 (DHEW/NIOSH Pub. No.
5.2. National Institute for Occupational Safety and Health:
Method No. S317, in NIOSH Manual of Analytical Methods. 2nd
ed., Vol. 3 (DHEW/NIOSH Pub. No.
5.3. Thomsen, E. and R.M. Stern: A Simple Analytical
Technique for the Determination of Hexavalent Chromium in Welding
Fumes and Other Complex Matrices. Scand. J. of Work, Environ. and
Health
5.4. Molina, D. and M.T. Abell: An Ion Chromatographic
Method for Insoluble Chromates in Paint Aerosol. Am. Ind. Hyg.
Assoc. J.
5.5. Vitale, R.J., G.R. Mussoline, J.C. Petura, and B.R.
James: Hexavalent Chromium Extraction from Soils. J. Environ.
Qual.
5.6 Zatka,V.J.: Speciation of Hexavalent Chromium in Welding
Fumes Interference by Air Oxidation of Chromium. Am. Ind. Hyg.
Assoc. J.
5.7. National Institute for Occupational Safety and Health:
Method No. 7600, in NIOSH Manual of Analytical Methods. 3rd ed.
(DHHS/NIOSH Pub. No.
5.8. Occupational Safety and Health Administration Salt Lake
Technical Center: Hexavalent Chromium (USDOL/OSHA Method
No.
5.9. Abell, M.T. and J.R. Carlberg: A Simple Reliable Method
for the Determination of Airborne Hexavalent Chromium. Am. Ind.
Hyg. Assoc. J.
5.10. Occupational Safety and Health Administration Salt Lake
Technical Center: Hexavalent Chromium Backup Data Report by
J.C. Ku
5.11. Dutkiewicz, R., J. Konczalik, and M. Przechera:
Assessment of the Colorimetric Methods of Determination of Chromium in
Air and Urine by Means of Radioisotope Techniques. Acta Pol. Pharm.
5.12. Dionex Corporation: Determination of Cr(VI) in Water, Wastewater, and Solid Waste Extracts, Technical Note 26. Sunnyvale, CA, 1990.
5.13. National Institute for Occupational Safety and Health:
Backup Data Report, Chromic Acid and Chromates, No. S317, in
Documentation of the NIOSH Validation Tests by D. Taylor, R.
Kupel, and J. Bryant (DHEW/NIOSH Pub. No.
5.14. Occupational Safety and Health Administration Salt Lake Technical Center: Quality Control Data - Chromate Analysis by B. Babcock. Salt Lake City, UT, 1982 - 1989.
5.15. Manufacturing Chemists Association: Properties and
Essential Information for Safe Handling and Use of Chromic Acid and
Chromates. (Chemical Safety Data Sheet
5.16. U.S. Department of Health and Human Services:
Update Toxicological Profile for Chromium
5.17. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Chromium, Nickel and Welding: Volume 49, International agency for Research on Cancer, Secrtariat of the World Health Organization:UK, 1990, ISBN 92 832 1249 5.
5.18 Harper, M.: SKC Inc., Information regarding using
the
5.19. Occupational Safety and Health Administration Salt Lake Technical Center: Ion Chromatography Standard Operating Procedure (Ion Chromatographic Committee). Salt Lake City, UT. In progress.
5.20. National Institute for Occupational Safety and Health:
Criteria for a Recommended Standard - Occupational Exposure to
Cr(VI) (DHEW/NIOSH Pub. No.
5.21. Mandel, J.: Accuracy and Precision, Evaluation and
Interpretation of Analytical Results, The Treatment of Outliers. In
Treatise On Analytical Chemistry. 2nd ed., Vol. 1, edited by I.
M. Kolthoff and P. J. Elving. New York: John Wiley and Sons, 1978. pp.
5.22. National Institute for Occupational Safety and Health:
Documentation of the NIOSH Validation Tests by D. Taylor, R.
Kupel, and J. Bryant (DHEW/NIOSH Pub. No.
5.23. Occupational Safety and Health Administration Salt Lake
Technical Center: Evaluation Guidelines of the Inorganic Methods
Branch. In OSHA Analytical Methods Manual. 2nd ed. Cincinnati,
OH: American Conference of Governmental Industrial Hygienists, 1991.
pp.
5.24. Burkart, J.A.: General Procedures for Limit of
Detection Calculations in the Industrial Hygiene Chemistry Laboratory.
Appl. Ind. Hyg.
5.25. National Institute for Occupational Safety and Health: Standard Operating Procedures for Industrial Hygiene Sampling and Chemical Analysis (SOP 018) Cincinnati, OH: National Institute for Occupational Safety and Health, Revised Sept., 1992.